 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A fluorescent light-up probe as an inhibitor of intracellular β-tryptase†
Qi
Wang‡
a,
Xiuyin
Shi‡
b,
Xiaoxia
Zhu
a,
Martin
Ehlers
c,
Junchen
Wu
*d and
Carsten
Schmuck
*c
aCollege of Public Health, Nantong University, 9 Seyuan Road, Nantong, 226019, China
bMedical Laboratory Center, Affiliated Hospital of Nantong University, Nantong, 226019, China
cInstitute for Organic Chemistry, University of Duisburg-Essen, Universitatsstrasse 7, Germany. E-mail: carsten.schmuck@uni-due.de; Fax: +49 201 183 4259; Tel: +49 201 183 3097
dKey Lab for Advanced Materials and Institute of Fine Chemicals, East China University of Science and Technology, 200237, China. E-mail: jcwu@ecust.edu.cn; Fax: +86 21 6425 2258; Tel: +86 21 6425 3674
First published on 28th April 2014
Abstract
A pyrene-functionalized peptidic inhibitor 1 binds to and inhibits β-tryptase in a non-competitive and reversible manner even in cells. Upon protein binding a fluorescence increase of the two pyrene fluorophores is observed which allows using 1 as a fluorescent light-up probe for this enzyme.
Fluorescent probes have been widely used as essential tools for the detection and imaging of biological targets in modern chemical biology.1 For example, several pyrene-based fluorescent probes for enzymes functioning under physiological conditions were developed and applied in living cells and tissues.2 The advantage of pyrene fluorophores is their sensitivity to the hydrophobicity of their corresponding environment. Hence, their emission is intensified upon binding to hydrophobic regions of a protein (e.g. within pockets and clefts on the surface of an enzyme). Furthermore, when multiple pyrene fluorophores interact e.g. in a hydrophobic pocket of a protein, formation of excimers and exciplexes with significantly altered emission properties compared to a single pyrene fluorophore can be induced. From such fluorescence changes the quantitative analysis of the binding of such molecules to the protein is possible.3,4
We have recently shown5 that multivalent cationic peptide ligands inhibit the serine protease β-tryptase in a reversible and non-competitive way by binding to the surface of the protein. β-Tryptase6 is one of the most abundant secretory granule-derived serine proteinases in mast cells. It has been used as a marker for mast cell activation,7 is involved in allergenic responses and is furthermore suspected to act as a mitogen for fibroblast lines.8 The structure of β-tryptase is rather unique, as it is a tetramer composed of four identical monomers around a central pore. The four active sites (each within one monomer) are embedded within this central pore and therefore any substrate (or any competitive inhibitor) has to enter the pore to reach the active sites. The cationic ligands we developed bind to the rim of anionic residues around the entrance to that pore, blocking the access to the active sites, thus shutting down the enzyme. However, so far our studies were restricted to in vitro assays and kinetic analyses of the isolated enzyme using non-fluorogenic inhibitors.
In this work, we now show that we can extend this approach of enzyme inhibition by protein surface binding to fluorescent inhibitors which also function in live cells. The new peptide ligand 1 contains two Lys-Trp-Lys tripeptide units, each attached via the C-terminus to a central lysine (Scheme 1). The N-terminus of each peptide arm was further functionalized with a pyrene moiety. This lysine rich peptide should also be capable of binding to the negatively charged surface of β-tryptase and this binding should change the pyrene fluorescence due to changes in the conformation of 1. Synthesis of 1 was achieved using microwave-assisted solid-phase peptide synthesis (ESI†).
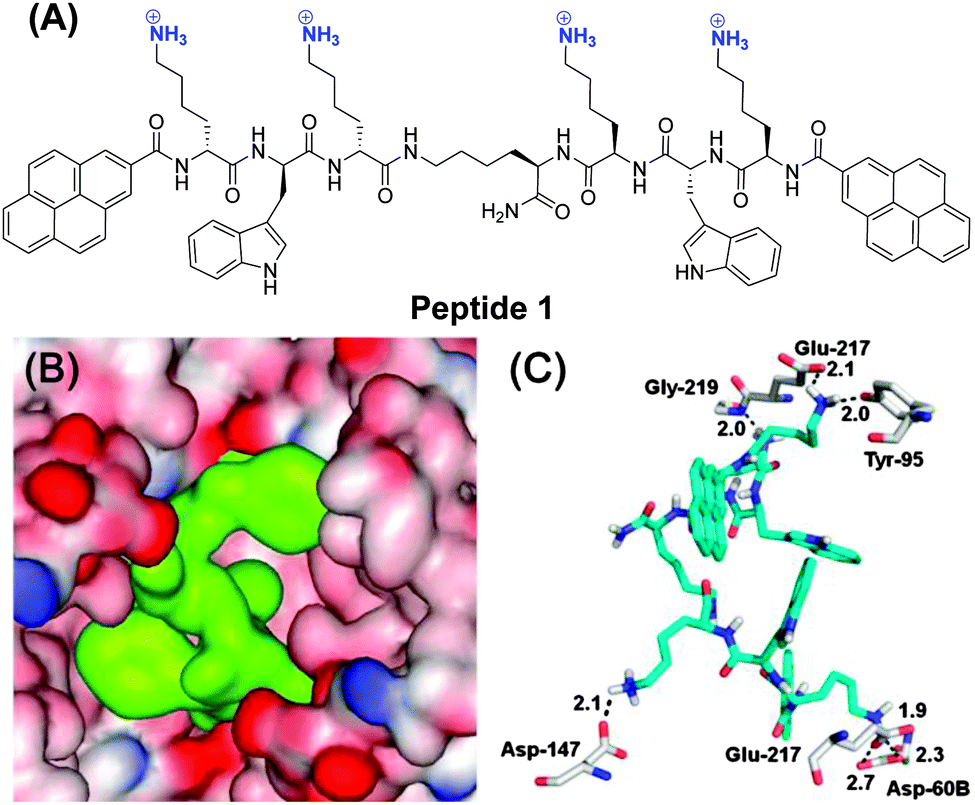 | ||
| Scheme 1 (A) Molecular structure of 1; (B) the surface of β-tryptase features clusters of negatively charged amino acids (red) around the entrance to the central pore in which the active sites are located. Ligand 1 (green) can bind to these negatively charged residues thereby filling the pore; (C) close-up of the binding interactions of 1 (cyan) with β-tryptase (grey) as obtained from a molecular modelling calculation. The cationic ammonium groups of 1 form ion pairs and hydrogen bonds with Gly-219, Glu-217, Tyr-95, Asp-60B, Asp-147 and Glu-217, respectively. | ||
Free 1 in solution shows a pyrene monomer emission at 400 nm and a weaker emission at 520 nm, characteristic of a pyrene excimer (Fig. 1), indicating that the two hydrophobic pyrene units in 1 are at least already partially stacked in the aqueous Tris-buffered saline (TBS) solution. Upon addition of β-tryptase from 0 to 20 nM, the excimer emission at 520 nm disappeared and the characteristic monomer emission at 400 nm increased significantly (Fig. 1A). This change in fluorescence is due to the increased hydrophobic microenvironment around the pyrene fluorophores and the restriction of their intramolecular rotation (RIR) within the complex (aggregation-induced emission, AIE).9 The hydrophobic microenvironment provided by the protein enhances the fluorescent emission of the pyrene fluorophores which is otherwise quenched in aqueous solution.
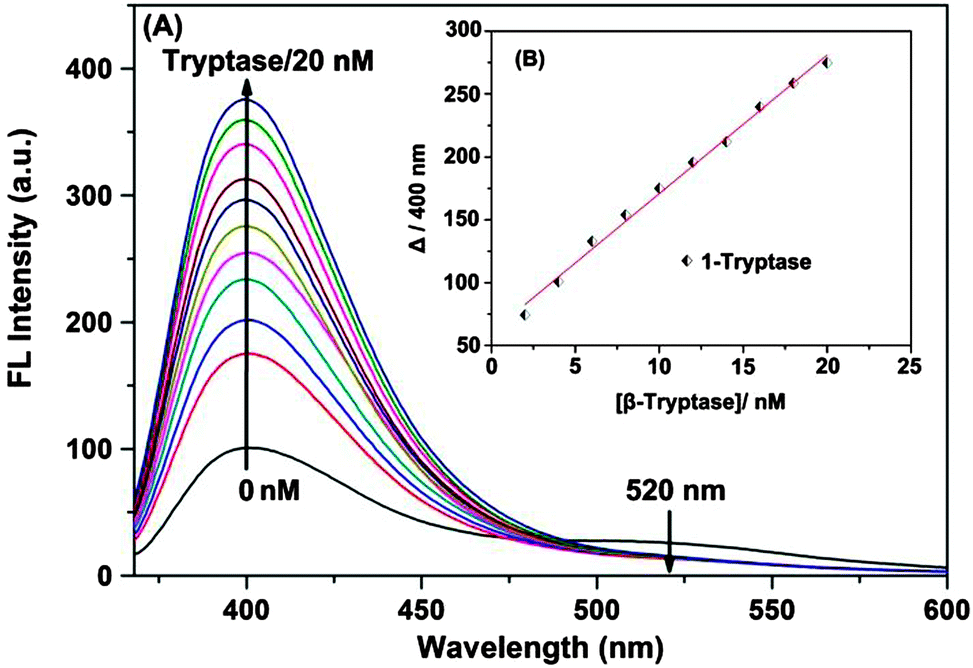 | ||
| Fig. 1 (A) Fluorescence response of an aqueous solution of 1 (500 nM) upon addition of β-tryptase (0–20 nM). (B) Inset: the corresponding titration curve showing the increase of the emission at 400 nm with increasing β-tryptase concentration. | ||
This change in fluorescence clearly shows that peptide ligand 1 binds to β-tryptase. Furthermore, the kinetic assay of enzymatic activity with rhSkin β-tryptase and Tos-Gly-Pro-Arg-AMC as a substrate confirmed that binding of 1 also inhibits the enzyme. Inhibition constants Ki were determined by incubating increasing concentrations of ligand 1 prior to determination of the residual enzyme activity as monitored by a standard assay (Table 1 and Fig. S4, ESI†). The ratio of reaction rates of inhibited to uninhibited substrate hydrolysis was then plotted versus the inhibitor concentration. From these data the apparent inhibition constants, Ki,app, were determined by nonlinear regression analysis using the equation: ν0/νi = 1 + [I]/Ki,app, where ν0 and νi are the uninhibited and inhibited reaction rates, respectively.10Ki values were obtained after correction for substrate competition according to Ki = Ki,app/(1 + [S]/Km)11 using the Michaelis constant for the substrate of Km = 300 μM, which was determined using substrate concentrations ranging from 0 to 800 μM (Fig. S3, ESI†). As the inhibition is reversible and non-competitive Ki and IC50 values are identical in this case. Accordingly, the determined IC50 values for inhibitor 1 are independent of the substrate concentration with IC50 = 40 ± 4 at [S] = 50 μM and 40 ± 2 at [S] = 100 μM, respectively. This clearly demonstrates that the inhibition is non-competitive (see also Fig. 2). With a Ki value of ca. 40 μM ligand 1 is a rather efficient inhibitor of β-tryptase, similar for example to the standard inhibitor p-Ab (Ki value: 57 μM) reported in literature.
| Enzyme | rhSkin β-tryptase | |
|---|---|---|
| The known literature standard inhibitor for β-tryptase para-aminobenz-amidine (p-Ab)12 has a Ki of 57 ± 8 μM. | ||
| [S] [μM] | 50 | 100 |
| IC50 [μM] | 40 ± 4 | 40 ± 2 |
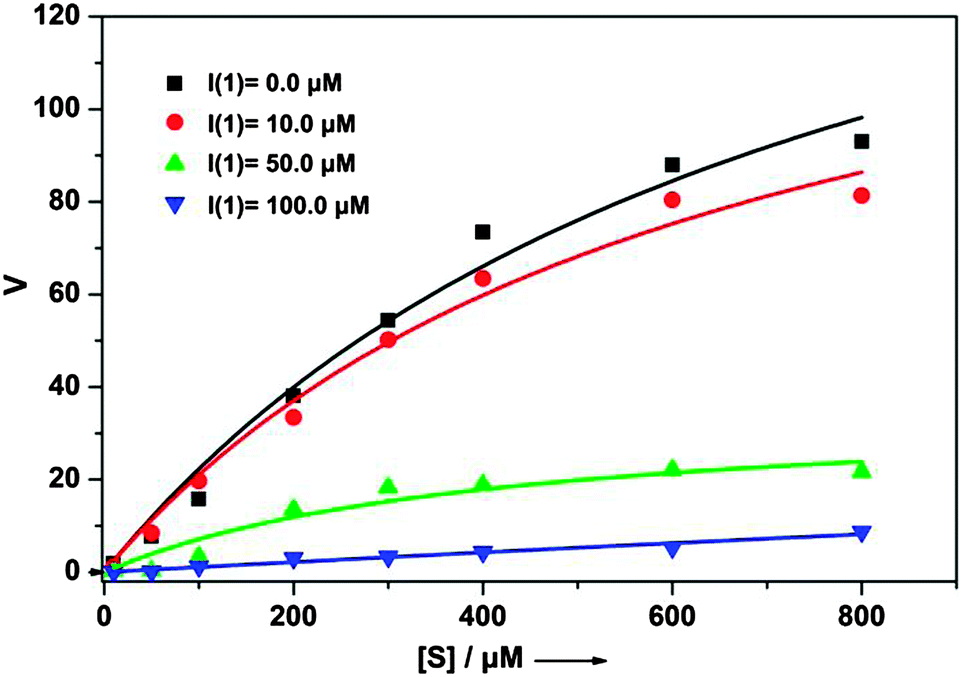 | ||
Fig. 2 Test for competitive versus non-competitive inhibition of ligand 1. With increasing inhibitor concentration [I], the maximum reaction rate (νmax) decreases whereas Km for the substrate is not changed, which indicates non-competitive inhibition ([I] = 0(■), 10 ( ), 50 ( ), 50 ( ), and 100 ( ), and 100 ( ) μM). ) μM). | ||
To fathom the selectivity of ligand 1 for β-tryptase, its binding affinity towards three other proteins was tested. Trypsin, chymotrypsin and bovine serum albumin (BSA), the first two of which are related serine proteases, are structurally similar to the corresponding β-tryptase monomer. Especially the active site of trypsin is highly similar to the active site of β-tryptase.13 However, as the data in Fig. S1 (ESI†) show, peptide 1 interacts significantly weaker with trypsin, chymotrypsin or BSA, at least under the conditions studied ([1] = 500 nM). Further studies confirmed that inhibition of β-tryptase by inhibitor 1 is reversible (enzyme activity can be restored after inhibition, Fig. S2, ESI†). To determine whether the inhibitor binds to the same binding site as the substrate (competitive inhibition) or to another site on the protein (non-competitive inhibition), the enzymatic activity at different substrate concentrations in the presence and absence of different amounts of inhibitor ([I] = 0, 10, 50, and 100 μM) was measured (Fig. 2). With increasing inhibitor concentration, the maximum reaction rate νmax for the enzymatic reaction significantly decreases even at high substrate concentration, whereas the Km-value for substrate binding (determined from the concentration at which the velocity ν is νmax/2) did not change. This clearly proves that ligand 1 is a non-competitive inhibitor as it is not in direct competition with the substrate for the same binding site.
A possible binding mode for 1 was derived from molecular modelling calculations. A docking study (for details see the ESI†) revealed the most stable binding mode that 1 binds to clusters of negatively charged amino acids on the surface of β-tryptase around the entrance to the central pore (Scheme 1B and C). The four cationic ammonium groups of 1 form ion pairs and hydrogen bonds with negatively charged residues, such as Glu-217, Asp-60B, Asp-147 and Glu-217, respectively. In this binding mode, ligand 1 nearly completely fills the central pore and thus prevents the substrate from reaching the active sites.
Interaction of 1 with β-tryptase was also tested in living mast cells (cell line CHMAS), which express significant amounts of β-tryptase, by measuring the effect of 1 on cell viability using a CCK-8 assay. As seen in Fig. 3A, ligand 1 effectively blocked β-tryptase in the CHMAS cell resulting in cell death. With increasing concentration of 1 the viability of the cells significantly decreased. At concentrations <0.5 μM, cell viabilities were larger than 70% after incubation for 24 h at 37 °C, whereas at concentrations >1 μM, cell viabilities dropped to <20% (IC50 = 0.7 μM; Fig. 3A, ESI†). Confocal laser scanning microscopy (CLSM) confirmed the presence of ligand 1 inside of the CHMAS cells (Fig. 3B and C). The effect of 1 on KB cells, which do not express large amounts of β-tryptase, was one order of magnitude weaker (IC50 = 6 μM), suggesting that indeed inhibition of β-tryptase is important for the effect of 1 on the viability of the cells.
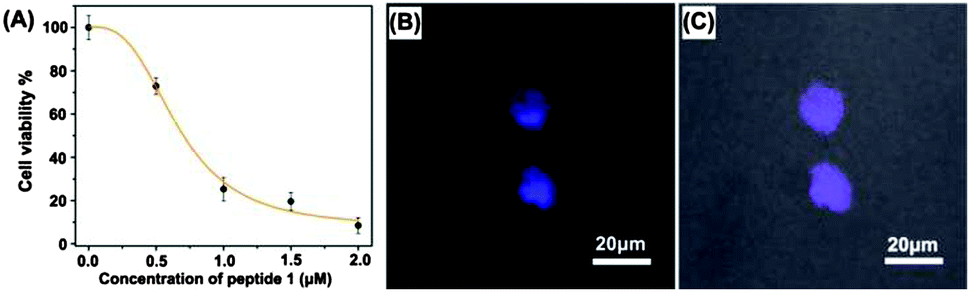 | ||
| Fig. 3 (A) Dose–response curve as measured by CCK-8 assays for 1 against mast cells (line CHMAS). The cells were seeded at 3 × 104 cells per well on a 96-well plate and incubated with various concentrations of peptide 1 (0.5–2.0 μM) for 24 h at 37 °C. The data are presented as mean ± SD (n = 3); confocal laser scanning microscopy (CLSM) images (B) of CHMAS cells treated with 10 μM peptide 1 for 30 min (channel: excitation: 404 nm, emission collected: 415–425 nm); (C) overlay of bright field and luminescence field images. | ||
In conclusion, we have developed a pyrene functionalized ligand 1 which inhibits β-tryptase in a reversible and non-competitive way most likely by protein surface binding. This leads to a strong increase in fluorescence as in the complex with the protein the pyrene moieties are in a more hydrophobic microenvironment with restricted internal rotational freedom. Hence, ligand 1 functions as a turn-on fluorescence sensor for β-tryptase. Furthermore, cell experiments indicate that 1 can be taken up by mast cells and exhibits inhibitory activity even within cells suppressing their growth. Therefore, ligand 1 may pave a way for applications in the treatment of allergenic response.
This work was funded by the National Basic Research 973 Program (2013CB733700) and the Fundamental Research Funds for the Central Universities (WJ1213007), the innovation program of shanghai municipal education commission (J100-2-13104), the Program of Shanghai Pujiang (K100-2-1275), the National Natural Science Foundation of China (21341001), Natural Science Foundation of Nantong University (12ZY034) and the German Science Foundation (DFG) (SFB 1093).
Notes and references
- (a) G. P. Drummen, Molecules, 2012, 17, 14067 CrossRef CAS PubMed; (b) T. N. N. Van and M. C. Morris, Prog. Mol. Biol. Transl. Sci., 2013, 113, 217 CrossRef PubMed; (c) Y. Hori, T. Norinobu, M. Sato, K. Arita, M. Shirakawa and K. Kikuchi, J. Am. Chem. Soc., 2013, 135, 12360 CrossRef CAS PubMed; (d) M. Kawaguchi, T. Okabe, S. Okudaira, K. Hanaoka, Y. Fujikawa, T. Terai, T. Komatsu, H. Kojima, J. Aoki and T. Nagano, J. Am. Chem. Soc., 2011, 133, 12021 CrossRef CAS PubMed.
- (a) J. Zheng, J. S. Li, Y. Jiang, J. Y. Jin, K. M. Wang, R. H. Yang and W. H. Tan, Anal. Chem., 2011, 83, 6586 CrossRef CAS PubMed; (b) A. Fujii, S. Hirota and T. Matsuo, Bioconjugate Chem., 2013, 24, 1218 CrossRef CAS PubMed; (c) J. Liu, J. Shi, J. Li and X. Yuan, Enzyme Microb. Technol., 2011, 49, 360 CrossRef CAS PubMed.
- Y. N. Hong, L. M. Meng, S. J. Chen, C. W. T. Leung, L. T. Da, M. Faisal, D. A. Silva, J. Z. Liu, J. W. Y. Lam, X. H. Huang and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 1680 CrossRef CAS PubMed.
- D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441 CrossRef CAS PubMed.
- (a) P. R. Wich and C. Schmuck, Angew. Chem., Int. Ed., 2010, 49, 4113 CrossRef CAS PubMed; (b) Q. Q. Jiang, L. Bartsch, W. Sicking, P. R. Wich, D. Heider, D. Hoffmann and C. Schmuck, Org. Biomol. Chem., 2013, 11, 1631 RSC.
- (a) D. N. Gosalia, C. M. Salisbury, J. A. Ellman and S. L. Diamond, Mol. Cell. Proteomics, 2005, 4, 626 CrossRef CAS PubMed; (b) N. Chen, J. Zou, S. M. Wang, Y. M. Ye, Y. Huang, G. Gadda and J. J. Yang, Biochemistry, 2009, 48, 3519 CrossRef CAS PubMed.
- (a) T. Tanaka, B. J. McRae, K. Cho, R. Cook, J. E. Fraki, D. A. Johnson and J. C. Powers, J. Biol. Chem., 1983, 258, 13552 CAS; (b) I. T. Harvima, N. M. Schechter, R. J. Harvima and J. E. Fräki, Biochim. Biophys. Acta, 1988, 957, 71 CrossRef CAS.
- (a) J. F. Molinari, M. Scuri, W. R. Moore, J. Clark, R. D. Tanaka and W. M. Abraham, Am. J. Respir. Crit. Care Med., 1996, 154, 649 CrossRef CAS PubMed; (b) W. M. Abraham, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2002, 282, 193 Search PubMed.
- (a) Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC; (b) Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- R. Lottenberg, J. A. Hall, J. W. d. Fenton and C. M. Jackson, Thromb. Res., 1982, 28, 313 CrossRef CAS.
- G. Sa R. J. Beynon and J. S. Bond, Proteolytic enzymes: a practical approach, IRL Press at Oxford University Press, 1989, p. 83 Search PubMed.
- T. Selwood, H. Smolensky, D. R. McCaslin and N. M. Schechter, Biochemistry, 2005, 44, 3580 CrossRef CAS PubMed.
- M. J. Costanzo, S. C. Yabut, H. R. Almond Jr., P. Andrade-Gordon, T. W. Corcoran, L. de Garavilla, J. A. Kauffman, W. M. Abraham, R. Recacha, D. Chattopadhyay and B. E. Maryanoff, J. Med. Chem., 2003, 46, 3865 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization data for peptide 1, and its absorption and emission data; enzyme activity assays, cell imaging data, and cell viability data. See DOI: 10.1039/c4cc02208d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |