 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic hydroxylation of benzene and toluene by an iron complex bearing a chelating di-pyridyl-di-NHC ligand†
Andreas
Raba
,
Mirza
Cokoja
,
Wolfgang A.
Herrmann
* and
Fritz E.
Kühn
*
Chair of Inorganic Chemistry/Molecular Catalysis, Catalysis Research Center, Technische Universität München, Ernst-Otto-Fischer-Straße 1, D-85747 Garching bei München, Germany. E-mail: wolfgangherrmann@ch.tum.de; fritz.kuehn@ch.tum.de; Fax: +49 89 289 13473; Tel: +49 89 289 13096
First published on 20th May 2014
Abstract
This work reports on iron-catalysed hydroxylation of benzene and toluene using aqueous H2O2. While benzene is hydroxylated with a high selectivity to phenol, toluene is hydroxylated to cresols with a high selectivity for the ortho and para-position. An inverse KIE indicates the presence of a high valent Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species during catalysis.
O species during catalysis.
The selective direct oxidation of aromatic compounds is among the most challenging reactions in organic synthesis.1 For more than a decade, this reaction has received considerable attention, since many modern polymers, pharmaceuticals, agrochemicals and biological active molecules bear hydroxyl-arene motifs, which are often synthesized in multi-step reactions.2 Even the simplest hydroxyl-arene, phenol, is currently being synthesized to a million-ton scale via a multi-step process with acetone as equimolar byproduct.3 Therefore it is not surprising that the oxidation of benzene by molecular oxygen has been defined as one of the “10 challenges in catalysis”.4 In nature, the biocatalytic oxygen transfer to arenes is achieved by oxygenase enzymes.5 Most of them contain one or two iron atoms in the active site, with cytochrome P450 being amongst the most prominent and best-studied ones.6 Several reports on iron model complexes as catalysts for arene oxidation have been published over recent years.7 In these oxidation reactions, an excess of substrate is often used to prevent overoxidation to the dihydroxylated product, and reactions are carried out at ambient temperatures with relatively low turnover numbers.
In the context of biomimetic model complexes, we have recently reported a series of organometallic FeII complexes with structural variations in the ligand sphere.8 Those complexes vary from the usual models, as the iron center is in part ligated by N-heterocyclic carbenes (NHCs) instead of chelating N, S, or O-donor ligands.9 The use of NHCs as ligands in transition metal catalysis has proven to be fruitful, as NHCs are excellent σ-donors compared to other donor ligands and exhibit high kinetic stability in the complexes.10 Furthermore, NHCs have successfully been applied in oxidation catalysis, exhibiting a higher stability towards ligand oxidation than is the case for phosphines.11
The Fe complex 1 bearing a tetradentate NCCN ligand8 is stable in air and water and has been shown to be an active catalyst for the epoxidation of olefins using aqueous hydrogen peroxide as the oxidant.12 In this study we set out to investigate the catalytic activity of 1 towards the oxidation of arenes to phenols. Adapting to previous reports on arene hydroxylation, complex 1 is used as the catalyst and H2O2 (30% in H2O) as the oxidant at 25 °C under air (Table 1). In a typical reaction, procedure complex 1 is dissolved in MeCN, benzene (100 equiv.) is added and the reaction is started with a single addition of aqueous H2O2 (30% in H2O, 100 equiv.). After 1 h, the reaction is quenched with an excess of PPh3, an external standard is added and the reaction solution is analysed by GC-FID (see the ESI†).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :benzene:H2O2 :benzene:H2O2 |
C [%] | S [%] | Y [%] |
| Reaction conditions: 5 mg of 1 in 2 mL of MeCN (3.2 mM), 61 μL benzene (100 equiv.), 70 μL H2O2 (100 equiv.); reaction time: 1 h; 25 °C; reaction is started with the addition of H2O2.a Conversion of benzene.b Selectivity for phenol.c Yield of phenol.d In 1 mL of acetonitrile (6.4 mM).e In 4 mL of acetonitrile (1.6 mM). | ||||
| 1 | 1:100:100 |
7.4 | 93.6 | 6.9 |
| 2 | 1:100:50 |
6.0 | 94.0 | 5.6 |
| 3 | 1:100:200 |
8.5 | 93.1 | 7.9 |
| 4 | 1:100:500 |
8.8 | 91.2 | 8.0 |
| 5 | 1:100:1000 |
13.0 | 87.0 | 11.2 |
| 6d | 1:100:100 |
10.2 | 89.2 | 9.1 |
| 7e | 1:100:100 |
5.9 | 93.6 | 5.5 |
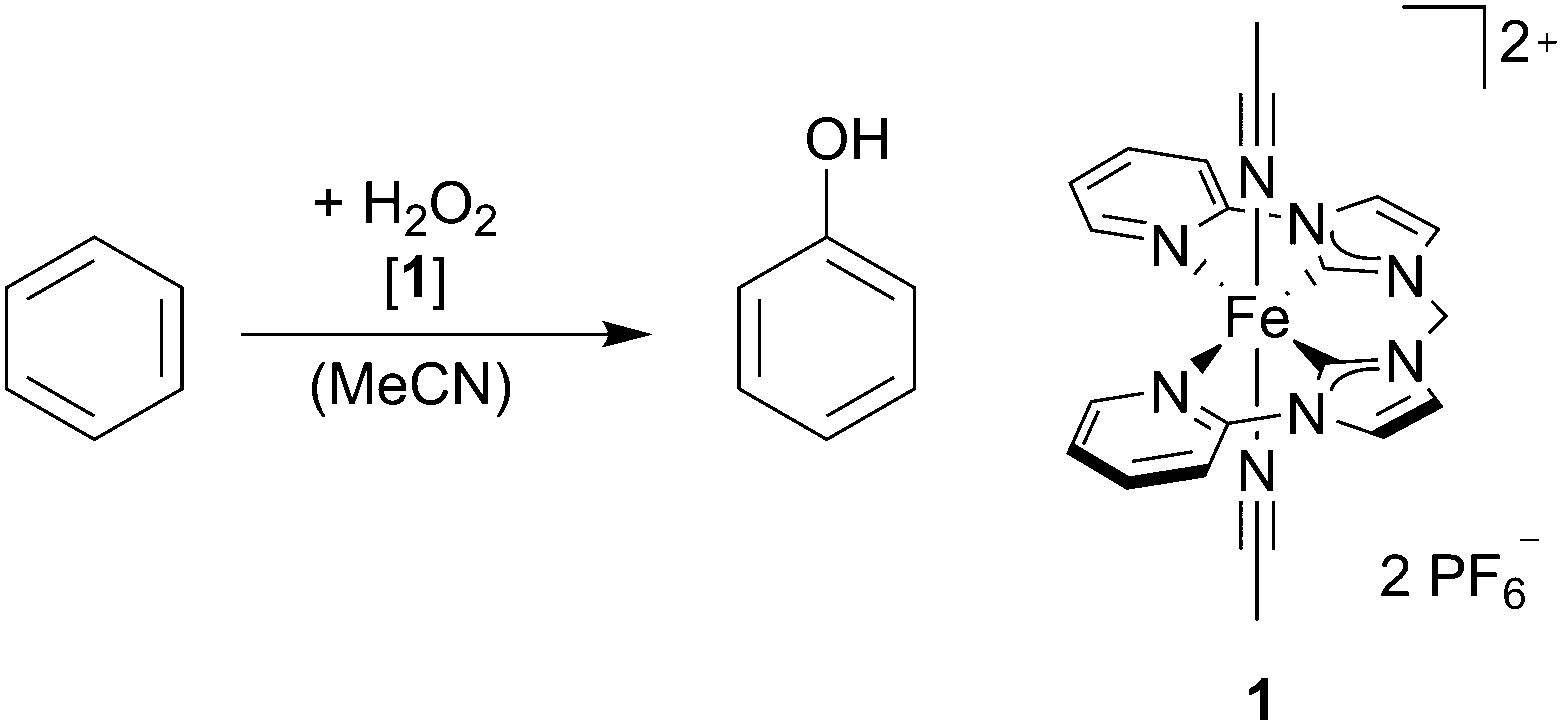
Although equimolar amounts of H2O2 and benzene are used, the conversion of benzene was 7.4% with a selectivity of 93.6% for phenol (Table 1, entry 1), whereas p-quinone was determined as the major byproduct by GC-MS/FID. In comparison to previously published reports on benzene hydroxylation, complex 1 lies so far within the range of the best systems in homogeneous phase.7a–c Although stoichiometric reactants are used with 1 mol% of the catalyst, a very high selectivity for phenol formation (>90%) is achieved. The influence of the stoichiometry on the catalytic reaction was investigated as shown in Table 1 (entries 1–5). Similar selectivities are observed when the molar ratio of benzene to H2O2 is changed from 1:2 to 5:1. Only when a high excess of H2O2 is applied the selectivity decreases substantially (Table 1, entry 5). Regarding the conversion, only a slight influence caused by the amount of H2O2 is observed. These results indicate that a defined reaction takes place. However, the catalyst suffers deactivation over time as proven by a second addition of H2O2, resulting in no significant change on the product yield. Interestingly, the molar concentration of 1 is relevant for the conversion (Table 1, entries 6 and 7). With a higher concentration, the conversion increases to 10%, whereas a lower concentration leads to a lower conversion. This is in accordance with the hypothesis of catalyst deactivation, as the substrate oxidation becomes less likely at lower concentrations and the decay of a potentially short lived intermediate might lead to catalyst deactivation. The temperature was further investigated as a factor influencing the catalyst lifetime, showing the following clear tendency: at higher temperatures the selectivity decreases drastically, however, the conversion is significantly increased, which means that overoxidation becomes dominant at higher temperatures (see the ESI†). A radical mechanism cannot be entirely excluded, as a Fenton reaction might take part in the oxidation.13 Despite this observation, very high selectivities were observed at lower temperatures, which is in agreement with a defined reaction.
There are only few reports dealing with catalytic non-heme iron-mediated aromatic hydroxylation in the presence of peroxides.7a,b,d,14 Several iron species have been considered responsible for selective aromatic hydroxylation. While FeIII–OOH and Fe–OO−˙ have been excluded from theoretical considerations and experimental data,14b,c ˙OH radicals in a Fenton reaction have been proven to oxidize benzene.13 Nonetheless, in a free radical mechanism, equimolar amounts of biphenyl are produced due to a recombination reaction. Notably, such a byproduct was not observed. More likely, recent publications reveal that FeIVO or FeVO are the best candidates for aromatic hydroxylation.7d,14a,b,e Therefore two reaction pathways are considered, either an H-atom abstraction or an electrophilic attack (Scheme 1). The dominance of one pathway strongly depends on the character of the FeO unit, which is defined by the oxidation state, spin state and the first coordination sphere.14a,b In general, both pathways should be possible, however, experimental and theoretical investigations favour an electrophilic attack of FeO on the π-system of the arene. This is a good experimental indicator to elucidate whether an electrophilic substitution or an H-abstraction mechanism is the kinetic isotope effect (KIE). In the case of H-atom abstraction, a rather high KIE is expected due to the different bond energies of deuterated or protic educts, whereas a rather low KIE is expected in the case of an electrophilic attack. Experimentally, an inverse KIE was reported by some groups, which is assigned to sp2-to-sp3 hybridization change during the attack of FeO on the sp2 hybridized ring to form a σ-complex.7a,b,d In the case of complex 1, we determined an intramolecular KIE through competition experiments between perdeuterated and protic benzene of 0.9 (see the ESI†) indicating an electrophilic attack by an FeO species. However, despite all efforts, such a species could so far not be isolated. In order to obtain further experimental insight into the mechanism, toluene was selected as a substrate for several reasons. The bond dissociation energies differ in the aromatic and aliphatic positions, and therefore a different reactivity concerning the mechanism is expected (C–H-abstraction vs. electrophilic attack) (∼370 kJ mol−1 for CH2–H; ∼475 kJ mol−1 for p-H-C6H4Me).15 In the case of an electrophilic attack, the substrate should be more reactive and clear selectivities should be visible when adapting the concept of electrophilic aromatic substitution (EAS) from organic chemistry. Moreover, toluene is unfunctionalised and no precoordination can occur defining the regioselectivity and influencing the reactivity.
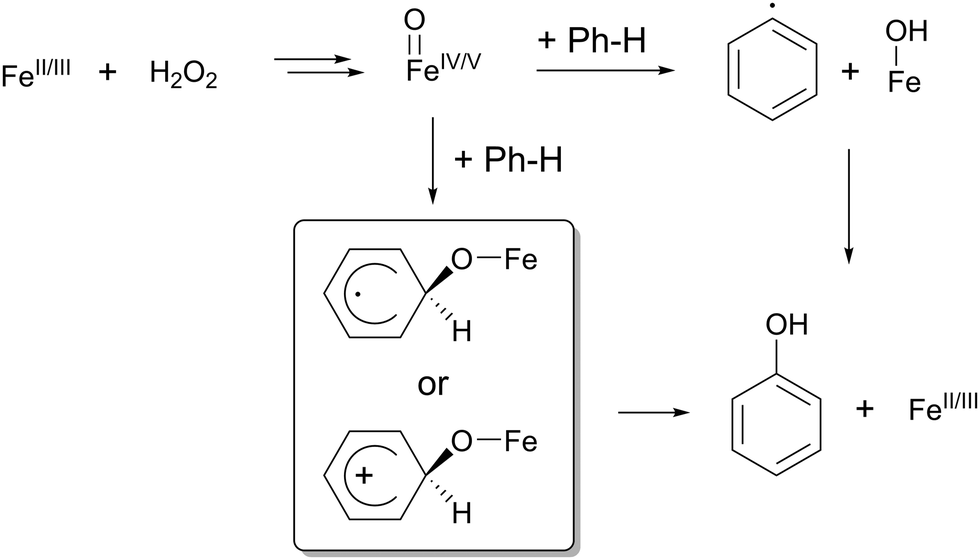 | ||
| Scheme 1 Possible reaction pathways for iron mediated aromatic hydroxylation. | ||
Catalyst 1 converts toluene in the presence of H2O2 in 15.2% to o/m/p-cresol as main products and benzaldehyde and benzyl alcohol as minor products (see Table 2). The high selectivity for ring oxidation of 77.9% clearly indicates an electrophilic mechanism by an FeO species during the catalytic reaction, whereas aliphatic oxidation is less likely (17.1%), as would be the case in a radical reaction or in H-atom abstraction. The residual 8.1% are mainly attributed to overoxidation to 2-methyl-p-quinone as identified by GC-MS/FID. Furthermore, the ortho- or para-position is clearly preferred, which can be adapted to the concept of EAS due to the directing effect of the methyl group (Table 2, entry 1). The ratio between ortho- and para-hydroxylation is about 1.6, which is slightly below the expected stochastic value of 2, and can be attributed to steric effects. The higher conversion of toluene compared to benzene is also expected for an EAS mechanism, as the aromatic system in toluene is more electron rich. Compared to benzene hydroxylation, the conversion behaves in a similar manner for toluene. Only a slight increase in the toluene oxidation is observed, going from 50 to 1000 equiv. of H2O2 (Table 2, entries 1–5). Regarding the selectivity, a tendency to side chain oxidation is observed with higher H2O2 concentration. However, the selectivity for aromatic hydroxylation is still 74.8% when 1000 equiv. H2O2 are applied. Interestingly, the formation of a meta-substituted product is increased with higher amounts of the oxidant. The molar concentration of the catalyst was further investigated, and best results and selectivities were obtained with a higher concentration (Table 2, entry 6).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry |
1:toluene:H2O2 or temperature |
Selectivity [%] | C [%] |
S
ring
[%] |
||||
| o-Cres. | m-Cres. | p-Cres. | Benzyl alcohol | Benzaldehyde | ||||
| Reaction conditions: 5 mg of 1 in 2 mL of MeCN (3.2 mM), 73 μL toluene (100 equiv.), 70 μL H2O2 (30% in H2O, 100 equiv.) for a catalyst:toluene:H2O2 ratio of 1:100:100; reaction time: 1 h; 25 °C; reaction is started with the addition of H2O2; for variations amounts are adapted; o/m/p-cresol are abbreviated as o/m/p-cres.a C: conversion of toluene.b Sring: selectivity for ring hydroxylation (mmol o/m/p-cresol/mmol converted toluene).c In 1 mL of acetonitrile (6.4 mM).d In 4 mL of acetonitrile (1.6 mM). |
||||||||
| 1 | 1:100:100 |
43.3 | 7.7 | 26.8 | 9.7 | 7.4 | 15.2 | 77.9 |
| 2 | 1:100:50 |
43.5 | 7.5 | 28.1 | 9.4 | 5.9 | 14.7 | 79.1 |
| 3 | 1:100:200 |
44.5 | 8.3 | 24.9 | 10.0 | 7.5 | 15.9 | 77.7 |
| 4 | 1:100:500 |
42.3 | 10.3 | 23.7 | 14.0 | 7.5 | 16.2 | 76.2 |
| 5 | 1:100:1000 |
38.9 | 12.4 | 23.5 | 12.1 | 8.5 | 19.7 | 74.7 |
| 6c | 1:100:100c |
48.0 | 7.8 | 25.6 | 7.3 | 6.0 | 14.7 | 81.4 |
| 7d | 1:100:100d |
32.8 | 9.2 | 26.7 | 16.0 | 11.7 | 14.3 | 68.7 |
| 8 | 60 °C | 18.9 | 6.6 | 16.0 | 23.9 | 14.2 | 12.8 | 41.5 |
| 9 | 50 °C | 22.9 | 7.8 | 18.5 | 19.0 | 11.8 | 12.4 | 49.2 |
| 10 | 40 °C | 30.0 | 9.1 | 21.9 | 16.9 | 12.6 | 11.7 | 61.2 |
| 11 | 30 °C | 39.7 | 8.6 | 25.9 | 11.8 | 8.1 | 15.7 | 74.3 |
| 12 | 10 °C | 44.5 | 6.4 | 27.9 | 9.2 | 6.8 | 12.0 | 78.8 |
| 13 | 0 °C | 46.4 | 5.1 | 29.2 | 9.2 | 6.3 | 11.8 | 80.7 |
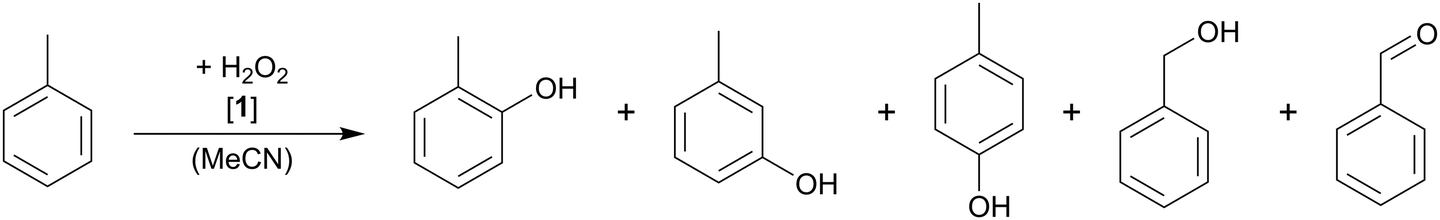
Furthermore, ring hydroxylation is clearly preferred at lower temperatures (Table 2, entries 1 and 8–13). At higher temperatures, oxidation at the aliphatic position becomes as likely as aromatic hydroxylation, which can either originate from a free radical reaction or from an H-atom-abstraction mechanism which is preferred at higher temperatures. The influence of the temperature on the conversion (highest at 30 °C) is presumably a consequence of both an increased catalyst deactivation and the kinetics of the reaction.
An inverse KIE of 0.8 was determined for toluene averaged on the aromatic positions, indicating again the electrophilic attack. Interestingly, an averaged KIE of 4.9 was determined for the oxidation at the aliphatic position, which is an indicator for an H-abstraction mechanism. In the case of a free radical mechanism, a lower KIE would be expected due to the high reactivity of ˙OH radicals.16 Furthermore, under those conditions no radical recombination products (e.g. bibenzyl) were observed.13 Comparing the results in toluene hydroxylation to other iron-based systems in the homogeneous phase, there are only few examples with high substrate excess and low TONs (<5).7b,f,g
In summary, we report the selective hydroxylation of benzene and toluene by an FeII NHC complex using aqueous H2O2 as oxidant. To the best of our knowledge, complex 1 is the most efficient homogeneous catalyst so far for the aromatic hydroxylation of toluene. The inverse KIE and the high regio- and chemoselectivity suggest that the mechanism of aromatic hydroxylation can be attributed to an electrophilic attack by a high valent FeO species. Comparatively high product yields were obtained in the hydroxylation of toluene, with selectivities for ring hydroxylation of higher than 80%, a total conversion of more than 15% at ambient conditions and a catalyst loading of 1 mol%. Although the total conversion is still to be improved, this report indicates some potential for future applications in organic synthesis, for example for fine chemicals.
Notes and references
- (a) D. A. Alonso, C. Najera, I. M. Pastor and M. Yus, Chem. – Eur. J., 2010, 16, 5274–5284 CrossRef CAS PubMed; (b) B. Lücke, K. V. Narayana, A. Martin and K. Jähnisch, Adv. Synth. Catal., 2004, 346, 1407–1424 CrossRef.
- J. H. Tyman, Synthetic and natural phenols, Elsevier, Amsterdam, 1996 Search PubMed.
- R. J. Schmidt, Appl. Catal., A, 2005, 280, 89–103 CrossRef CAS PubMed.
- B. Cornils and W. A. Herrmann, J. Catal., 2003, 216, 23–31 CrossRef CAS.
- R. Ullrich and M. Hofrichter, Cell. Mol. Life Sci., 2007, 64, 271–293 CrossRef CAS PubMed.
- P. R. Ortiz de Montellano, Cytochrome P-450 structure, mechanism, and biochemistry, Plenum Press, New York, 3 edn, 2010 Search PubMed.
- (a) A. Thibon, V. Jollet, C. Ribal, K. Senechal-David, L. Billon, A. B. Sorokin and F. Banse, Chem. – Eur. J., 2012, 18, 2715–2724 CrossRef CAS PubMed; (b) O. V. Makhlynets and E. V. Rybak-Akimova, Chem. – Eur. J., 2010, 16, 13995–14006 CrossRef CAS PubMed; (c) A. Thibon, J.-F. Bartoli, R. Guillot, J. Sainton, M. Martinho, D. Mansuy and F. Banse, J. Mol. Catal. A: Chem., 2008, 287, 115–120 CrossRef CAS PubMed; (d) S. P. de Visser, K. Oh, A.-R. Han and W. Nam, Inorg. Chem., 2007, 46, 4632–4641 CrossRef CAS PubMed; (e) S. Taktak, M. Flook, B. M. Foxman, L. Que, Jr. and E. V. Rybak-Akimova, Chem. Commun., 2005, 5301–5303 RSC; (f) D. Mathieu, J. F. Bartoli, P. Battioni and D. Mansuy, Tetrahedron, 2004, 60, 3855–3862 CrossRef CAS PubMed; (g) V. Balland, D. Mathieu, N. Pons-Y-Moll, J. F. Bartoli, F. Banse, P. Battioni, J.-J. Girerd and D. Mansuy, J. Mol. Catal. A: Chem., 2004, 215, 81–87 CrossRef CAS PubMed; (h) D. Bianchi, R. Bortolo, R. Tassinari, M. Ricci and R. Vignola, Angew. Chem., Int. Ed., 2000, 39, 4321–4323 CrossRef CAS.
- A. Raba, M. Cokoja, S. Ewald, K. Riener, E. Herdtweck, A. Pöthig, W. A. Herrmann and F. E. Kühn, Organometallics, 2012, 31, 2793–2800 CrossRef CAS.
- (a) W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed; (b) L. Que, Jr. and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed; (c) A. R. McDonald and L. Que, Coord. Chem. Rev., 2013, 257, 414–428 CrossRef CAS PubMed.
- H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687–703 CrossRef CAS PubMed.
- (a) T. Strassner, Top. Organomet. Chem., 2007, 22, 125–148 CrossRef CAS; (b) S. M. Podhajsky and M. S. Sigman, RSC Catal. Ser., 2011, 6, 345–365 CAS.
- J. W. Kück, A. Raba, I. I. E. Markovits, M. Cokoja and F. E. Kühn, ChemCatChem, 2014 DOI:10.1002/cctc.201402063R1.
- C. Walling and R. A. Johnson, J. Am. Chem. Soc., 1975, 97, 363–367 CrossRef CAS.
- (a) S. Sahu, L. R. Widger, M. G. Quesne, S. P. de Visser, H. Matsumura, P. Moënne-Loccoz, M. A. Siegler and D. P. Goldberg, J. Am. Chem. Soc., 2013, 135, 10590–10593 CrossRef CAS PubMed; (b) A. Ansari, A. Kaushik and G. Rajaraman, J. Am. Chem. Soc., 2013, 135, 4235–4249 CrossRef CAS PubMed; (c) R. Latifi, L. Tahsini, W. Nam and S. P. de Visser, Phys. Chem. Chem. Phys., 2012, 14, 2518–2524 RSC; (d) E. Olsson, A. Martinez, K. Teigen and V. R. Jensen, Chem. – Eur. J., 2011, 17, 3746–3758 CrossRef CAS PubMed; (e) M.-J. Kang, W. J. Song, A.-R. Han, Y. S. Choi, H. G. Jang and W. Nam, J. Org. Chem., 2007, 72, 6301–6304 CrossRef CAS PubMed; (f) T. Borowski, A. Bassan and P. E. M. Siegbahn, Inorg. Chem., 2004, 43, 3277–3291 CrossRef CAS PubMed.
- Y.-R. Luo, Comprehensive handbook of chemical bond energies, CRC Press, Boca Raton, 2007 Search PubMed.
- F. Gozzo, J. Mol. Catal. A: Chem., 2001, 171, 1–22 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed catalysis procedures, further catalysis results. See DOI: 10.1039/c4cc02178a |
| This journal is © The Royal Society of Chemistry 2014 |