 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aryloxymaleimides for cysteine modification, disulfide bridging and the dual functionalization of disulfide bonds†
Cristina
Marculescu
a,
Hanno
Kossen
a,
Rachel E.
Morgan
a,
Patrick
Mayer
a,
Sally A.
Fletcher
a,
Berend
Tolner
b,
Kerry A.
Chester
b,
Lyn H.
Jones
c and
James R.
Baker
*a
aDepartment of Chemistry, University College London, 20 Gordon St, London, UK. E-mail: j.r.baker@ucl.ac.uk; Tel: +44 (0)2076792653
bUCL Cancer Institute, 72 Huntley Street, London, WC1E 6BT, UK
cPfizer, Chemical Biology Group, BioTherapeutics Chemistry, WorldWide Medicinal Chemistry, 610 Main Street, Cambridge, MA 02139, USA
First published on 15th May 2014
Abstract
Tuning the properties of maleimide reagents holds significant promise in expanding the toolbox of available methods for bioconjugation. Herein we describe aryloxymaleimides which represent ‘next generation maleimides’ of attenuated reactivity, and demonstrate their ability to enable new methods for protein modification at disulfide bonds.
The maleimide motif 1 is a highly reactive conjugate acceptor and can be employed effectively in selective reactions with thiols, such as cysteine residues in proteins.1 As a result it is one of the most widely used functional groups in bioconjugation (Scheme 1). We have recently described bromomaleimides 3 as the parent members of a new class of reagents, which can be termed ‘next generation maleimides’ (NGMs). The incorporation of the leaving group leads to a switch in the mechanism that occurs upon reaction with a thiol, from an addition reaction to an addition–elimination reaction. The result is the formation of a thiomaleimide product 4, rather than thiosuccinimide 2. Such next generation maleimide reagents enable new applications including reversible cysteine modification,2 the construction of triconjugates,2b,3 and the bridging of disulfide bonds.2b,4
 | ||
| Scheme 1 Cysteine labelling of proteins using maleimide 1 and next generation maleimide (NGM) 3. | ||
We envisaged that the choice of leaving group on the maleimide core could result in reagents with finely tuned properties. For example the reactivity could be controlled, in a manner similar to the use of iodo-, bromo- and chloroacetamides as varyingly reactive reagents for selective cysteine modification.5 In addition to guiding the reactivity, the choice of leaving group could also control other properties of the reagents, such as aqueous solubility. Herein we report on the aryloxymaleimides, as NGMs of attenuated reactivity, and demonstrate application of these reagents in cysteine modification and in new modes of conjugation of disulfide bonds.
Having previously shown that thiophenol served as a suitable leaving group in dithiophenolmaleimide reagents,4b we postulated that phenol may do like-wise. Furthermore we considered that the increased mesomeric contribution from the oxygen into the maleimide ring would lead to reagents of reduced electrophilicity. The synthesis of aryloxymaleimides 6–8 was carried out by simple addition of the corresponding phenol to N-methyl bromomaleimide 5 (Scheme 2).
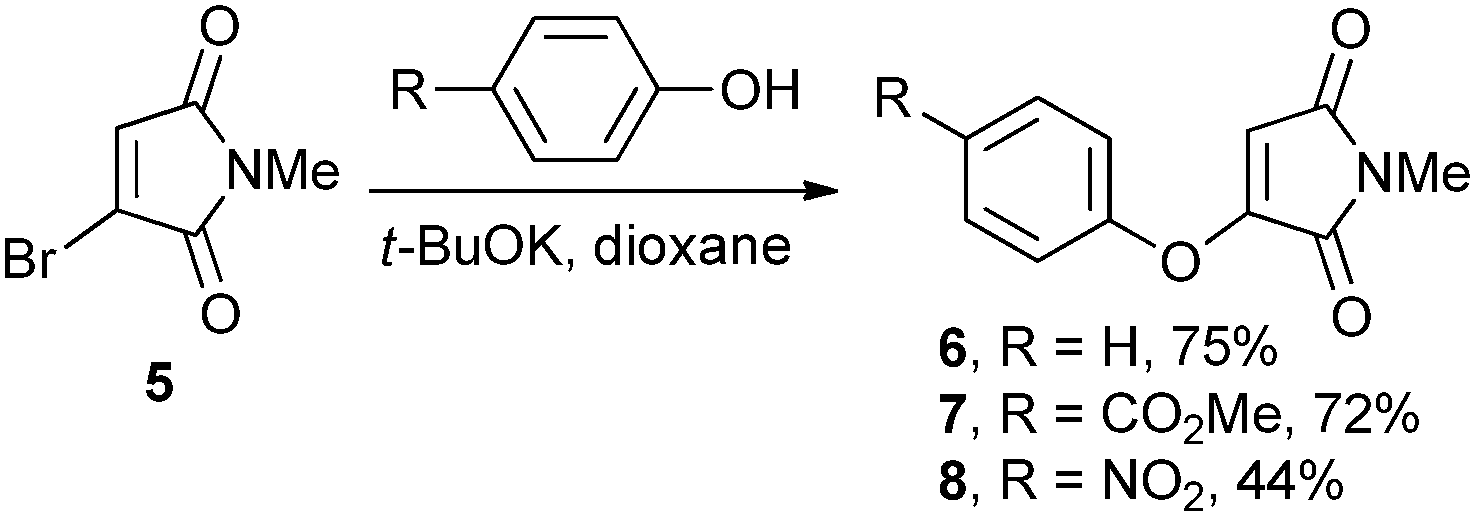 | ||
| Scheme 2 Synthesis of monoaryloxymaleimides 6–8. | ||
Addition of these reagents to a model protein, the single cysteine mutant (L111C) of the SH2 domain of the Grb2 adaptor protein 9,2b was monitored over 2 hours and compared to the use of bromomaleimide 5 (Fig. 1). All three aryloxymaleimides underwent the desired addition–elimination reaction to afford thiomaleimide conjugate 10, at a reduced rate compared to bromomaleimide 5.2b As expected, the incorporation of electron withdrawing groups on the aryloxy ring (7 and 8) led to an acceleration in rate compared to the unsubstituted phenoxymaleimide 6. It is notable that the aryloxymaleimides are still considerably more reactive than iodoacetamide, another commonly employed cysteine reactive reagent, which underwent <5% reaction with this protein after 30 min under the same conditions.2b Furthermore the reaction could be accelerated by increasing the number of equivalents of phenoxymaleimide 6 to ten, leading to complete conversion to the product in just 10 min. These results demonstrate that phenoxymaleimides represent cysteine modification reagents of attenuated reactivity, a feature we postulated could be useful in affording improved selectivity in certain bioconjugations.
 | ||
| Fig. 1 Addition of aryloxymaleimides to Grb2 SH2 (L111C), with a graph of conversion over time measured by LCMS. | ||
A particular regioselectivity problem had become apparent in our studies on disulfide bridging for which we envisaged that the aryloxymaleimides could offer an ideal solution. We were interested in attempting to use bromomaleimide to bridge a disulfide bond in the peptide hormone somatostatin 11, in a manner related to our work with dibromomaleimide.2b,4b This would afford a saturated dithiosuccinimide 14 (presumed to be as a mixture of diastereomers), in contrast to the dithiomaleimides generated by dibromomaleimide bridging. Such dithiosuccinimides would be expected to have different properties to dithiomaleimides (e.g. reduced reactivity with thiols)2b,3b and could serve as a complementary approach. However, addition of bromomaleimide 5 to reduced somatostatin 12 led to a mixture of the desired bridged adduct 14 and the bis-addition product 15 (Scheme 3). Thus addition of bromomaleimide to the second thiol in intermediate 13 was competing with the bridging reaction. What was required was an NGM analogue which underwent a slower reaction with cysteine, to ensure that the cyclisation of 13 would take place before the second maleimide addition. The aryloxymaleimides appeared to fit this specification. Indeed treatment of reduced somatostatin 12 with phenoxymaleimide 6 led after 1 h to quantitative conversion to the succinimide bridged somatostatin conjugate 14. Increasing the amount of phenoxymaleimide to 10 equiv. in this reaction still afforded none of the bis-labelled product 15, demonstrating the extremely high selectivity for disulfide bridging. To prove that succinimide bridging had reached completion, product 14 was treated with 1 equiv. of maleimide. No reaction was observed by LC-MS analysis after 5 min at room temperature, proving the absence of detectable amounts of free thiol.
 | ||
| Scheme 3 Succinimide bridging of the disulfide bond in somatostatin. | ||
We also found that, unlike bromomaleimides,4b aryloxymaleimides are resistant to cross-reactivity with the reducing agent TCEP and therefore that the succinimide bridging could be carried out in situ. The optimised in situ conditions involve addition of 1.5 equiv. aryloxymaleimide to the peptide followed by addition of the equimolar amount of the reducing agent TCEP leading to quantitative conversion after 2 h. In situ protocols are valuable in disulfide bridging as they limit the amount of time the peptide or protein lacks its bridging functionality and thus reduces the propensity to aggregate or unfold.4b,6
A selection of derivatised aryloxymaleimides (Fig. 2) were then synthesised (see ESI,† Scheme S1) to explore the scope of this succinimide bridging. Notably a new, scalable synthesis of bromomaleimide was developed to allow convenient access to these molecules. A water soluble aryloxy motif was incorporated in 16 to investigate the possibility of organic solvent free labelling. A ‘miniPEG’ phenoxymaleimide 17 was developed as a model for PEGylation of proteins, and rhodamine phenoxymaleimide 18 for fluorescent labelling.
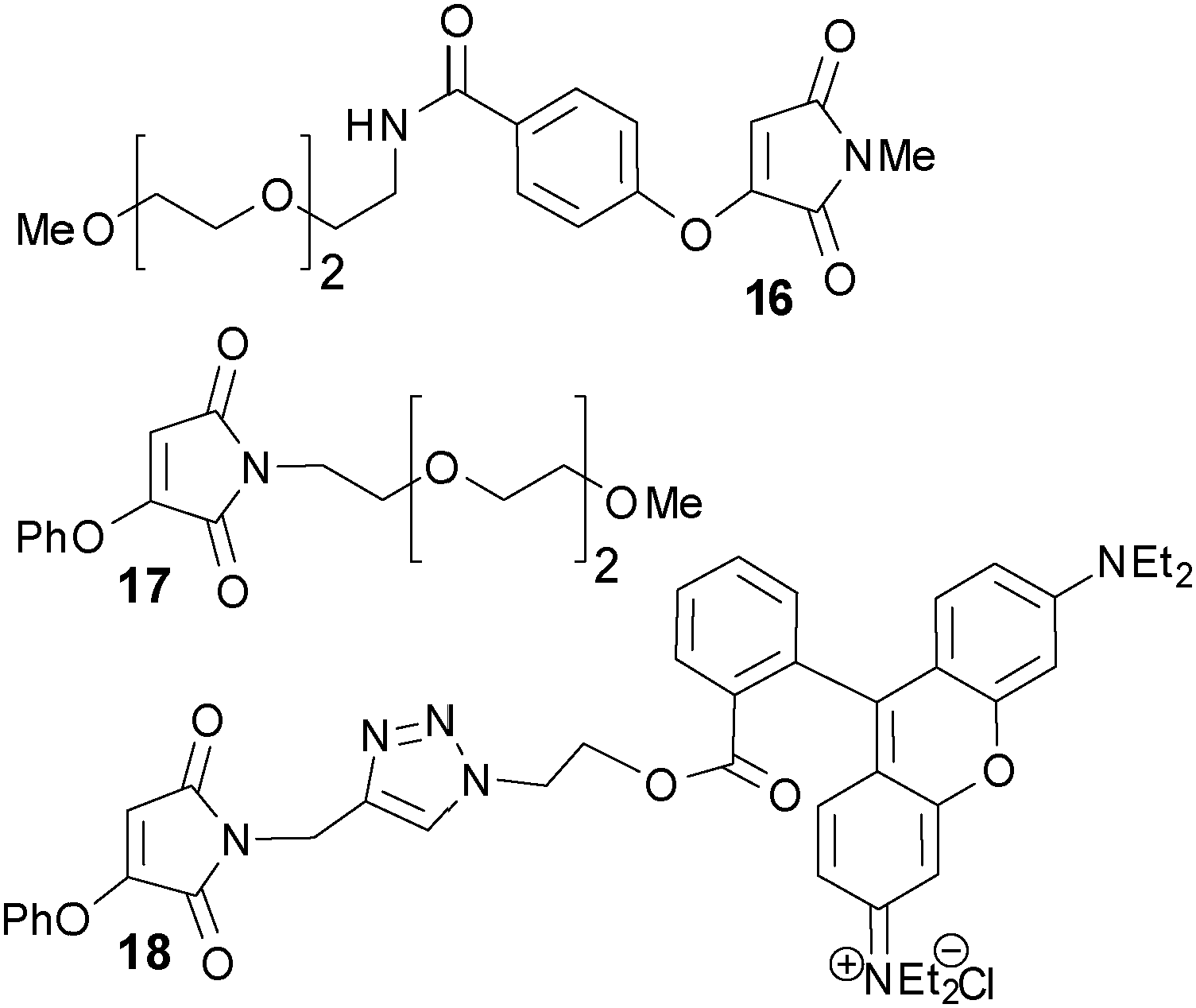 | ||
| Fig. 2 Aryloxymaleimide derivatives. | ||
The use of the water soluble aryloxymaleimide 16 successfully enabled the labelling of somatostatin under organic solvent free conditions. It is noteworthy that this is the first example of a maleimide reagent incorporating a water solubilising group which is released upon conjugation, and further exemplifies the enhanced versatility of NGMs over traditional maleimides.
To demonstrate that the succinimide bridging was also applicable to larger proteins, chemical modification of an antibody fragment was investigated. A substantial portion of all biotechnology products under development are antibodies or antibody fragments,7 and homogeneous conjugation technologies have a key role to play in realising the full potential in this area.6,8 Treatment of cystine stabilized anti-carcinoembryonic antigen single chain Fv fragment (anti-CEA ds-scFv) 196 with N-miniPEG phenoxymaleimide 17 and benzeneselenol as the reducing agent6 led to quantitative conversion to the conjugate 20 (Scheme 4). The same outcome was observe using N-methyl phenoxymaleimide 6 to afford conjugate 21.
 | ||
| Scheme 4 Succinimide bridging of ds-scFv shMFE. | ||
We have previously demonstrated that maleimide bridges are susceptible to thiol mediated cleavage at high concentration of thiols, such as the mM concentrations found in the cytoplasm of cells.2b,6 We wanted to test the stability of the succinimide bridge to thiols, to investigate if a different reactivity profile was demonstrated. Treatment of succinimide bridged scFv conjugate 21 with 100 equiv. GSH (7 mM, pH 7.4) led to no reaction even after 48 h at room temperature. Thus the succinimide bridge in this scFv is completely resistant to thiol mediated cleavage in contrast to the maleimide bridge.
Returning to somatostatin however, it became apparent that this thiol stability is variable depending on the exact nature of the disulfide bridge. A comparison of maleimide and succinimide bridged somatostatin revealed that both were cleaved at similar rates by GSH under cytoplasm mimicking conditions to release the free peptide 12 (see ESI,† Fig. S1). We propose that the mechanism for the thiol cleavage of the succinimide bridge starts with a retro-conjugate addition to generate acyclic thiomaleimide 13 (see Scheme 3), and thus 13 and 14 are in fact in equilibrium. Thiomaleimide 13 is then cleaved by GSH in a manner consistent with our previous results,9via a conjugate addition–elimination sequence. The difference in thiol stability of the somatostatin–succinimide 14 to the scFv-succinimide 20 we postulate to be attributable to the extent of intramolecular interactions that are present constraining these structures. In the case of somatostatin the formation of acyclic thiomaleimide intermediate 13 can be followed by a shift in conformation reducing the rate of rebridging and allowing a significant amount of this intermediate to form in solution. In contrast in the case of the scFv the extensive interactions between the variable regions of the light and heavy chain hold the cysteines in close proximity, precluding the formation of significant amounts the acyclic thiomaleimide intermediate required for thiol mediated cleavage.
Notably employing N-phenyl phenoxymaleimide to bridge somatostatin led to the resultant conjugate which underwent rapid hydrolysis of the succinimide bridge (consistent with the equivalent N-phenyl maleimide bridge,4a and see ESI,† Fig. S2), affording a succinamic acid which was completely stable to thiols; this represents a general method of producing completely thiol stable conjugates.
Whilst the focus of this study was investigating the use of phenoxymaleimides in succinimide bridging, we realised that the reversibility of the bridging offered an interesting possibility for dual labelling of disulfide bonds. We envisaged that addition of a second electrophile could lead to interception of intermediate 13, attaching a second tag. Dual labelling of a reduced disulfide bond is extremely difficult to achieve using traditional reagents as it relies on a significant difference in reactivity between the two cysteines.10 Methods for the site-selective dual-labelling of peptides are widely sought, to allow controlled introduction of multiple functionalities.11 For example, the incorporation of two different fluorophores allows FRET studies while the attachment of an in vivo stabilising group (e.g. a PEG) and a drug can prospectively enable therapeutic applications.
Thus treatment of reduced somatostatin 12 with phenoxymaleimide 6 (R1 = Me) afforded the succinimide bridged somatostatin 14 which was then treated, without purification, with monobromomaleimide (R2 = H) (Scheme 5). The reaction required warming to 37 °C for 2 h to ensure complete conversion to the dual modified peptide 22. To confirm the formation of two cleavable thiomaleimides the conjugate was treated with 2-mercaptoethanol (100 equiv.) to quantitatively convert to reduced somatostatin. To demonstrate that this approach could be employed to access dual functionalised peptides, we constructed miniPEG–fluorescein and rhodamine–fluorescein bis-conjugates (23 and 24). Notably other electrophiles can also be employed in the second modification step; for example maleimide was employed affording a dual conjugate in which one of the tags was cleavable by thiols and the other was not. Trypsin digest of 25 confirmed that the reaction led, as expected, to a mixture of the two possible regioisomers (see ESI,† Fig. S3).
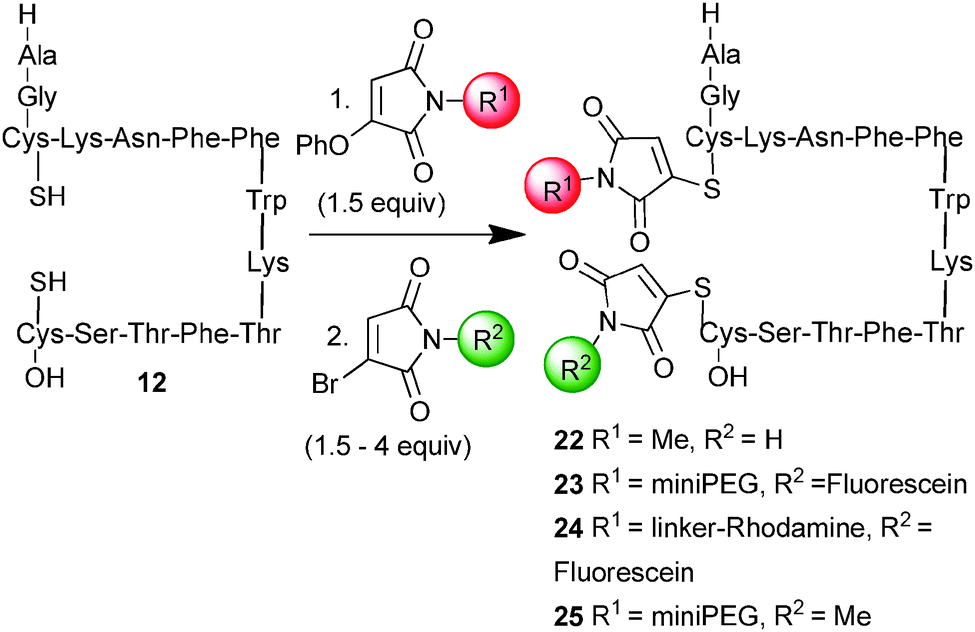 | ||
| Scheme 5 Dual modification of somatostatin (conditions: 1. 1 h, r.t., pH 6.4; 2. 2 h, 37 °C, pH 6.4). | ||
In conclusion, by varying the leaving groups on a maleimide core, a range of next generation maleimides can be produced with tuneable properties. Here a selection of aryloxymaleimides have been demonstrated to serve as efficient cysteine labelling reagents of attenuated reactivity. This enables the aryloxymaleimides to be employed in succinimide bridging, offering a novel strategy for disulfide labelling. The reversibility of the succinimide bridge in the peptide also enabled a one-pot dual labelling strategy to be developed.
The authors gratefully acknowledge Pfizer, BBSRC and UCL for supporting this work.
Notes and references
- (a) J. D. Gregory, J. Am. Chem. Soc., 1955, 77, 3922–3923 CrossRef CAS; (b) R. A. Bednar, Biochemistry, 1990, 29, 3684–3690 CrossRef CAS; (c) J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem. – Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- (a) L. M. Tedaldi, M. E. B. Smith, R. Nathani and J. R. Baker, Chem. Commun., 2009, 6583–6585 RSC; (b) M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- (a) J. Youziel, A. R. Akhbar, Q. Aziz, M. E. B. Smith, S. Caddick, A. Tinker and J. R. Baker, Org. Biomol. Chem., 2014, 12, 557 RSC; (b) P. Moody, M. E. B. Smith, C. P. Ryan, V. Chudasama, J. R. Baker, J. Molloy and S. Caddick, ChemBioChem, 2012, 13, 39–41 CrossRef CAS PubMed.
- (a) C. P. Ryan, M. E. B. Smith, F. F. Schumacher, D. Grohmann, D. Papaioannou, G. Waksman, F. Werner, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 5452–5454 RSC; (b) F. F. Schumacher, M. Nobles, C. P. Ryan, M. E. B. Smith, A. Tinker, S. Caddick and J. R. Baker, Bioconjugate Chem., 2011, 22, 132–136 CrossRef CAS PubMed; (c) M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 1847–1852 CrossRef CAS PubMed; (d) M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, Chem. Commun., 2012, 48, 4064–4066 RSC.
- R. L. Lundblad, Chemical reagents for protein modification, CRC Press, 2005 Search PubMed.
- F. F. Schumacher, V. A. Sanchania, B. Tolner, Z. V. F. Wright, C. P. Ryan, M. E. B. Smith, J. M. Ward, S. Caddick, C. W. M. Kay, G. Aeppli, K. A. Chester and J. R. Baker, Sci. Rep., 2013, 3, 1525 Search PubMed.
- S. Aggarwal, Nat. Rev. Drug Discovery, 2010, 9, 427–428 CrossRef CAS PubMed.
- M. M. Sun, K. S. Beam, C. G. Cerveny, K. J. Hamblett, R. S. Blackmore, M. Y. Torgov, F. G. Handley, N. C. Ihle, P. D. Senter and S. C. Alley, Bioconjugate Chem., 2005, 16, 1282–1290 CrossRef CAS PubMed.
- R. I. Nathani, V. Chudasama, C. P. Ryan, P. R. Moody, R. E. Morgan, R. J. Fitzmaurice, M. E. B. Smith, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2013, 11, 2408–2411 CAS.
- Y. Santoso, C. M. Joyce, O. Potapova, L. Le Reste, J. Hohlbein, J. P. Torella, N. D. F. Grindley and A. N. Kapanidis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 715–720 CrossRef CAS PubMed.
- R. Nathani, P. Moody, V. Chudasama, M. E. B. Smith, R. Fitzmaurice and S. Caddick, Chem. Sci., 2013, 4, 3455 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and full spectroscopic data are available. See DOI: 10.1039/c4cc02107j |
| This journal is © The Royal Society of Chemistry 2014 |