 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric synthesis of functionalized cyclohexanes bearing five stereocenters via a one-pot organocatalytic Michael–Michael–1,2-addition sequence†
Pankaj
Chauhan
,
Gregor
Urbanietz
,
Gerhard
Raabe
and
Dieter
Enders
*
Institute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany. E-mail: enders@rwth-aachen.de
First published on 15th May 2014
Abstract
A highly stereoselective one-pot procedure involving an enantioselective Michael addition promoted by low loading of an amino-squaramide catalyst followed by an achiral base catalyzed domino Michael–Knoevenagel-type 1,2-addition sequence provides efficient access to fully substituted cyclohexanes bearing five contiguous stereogenic centers in good yields (68–86%) and excellent stereoselectivities (>30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 96–99% ee).
:1 dr and 96–99% ee).
The asymmetric synthesis of complex molecular structures bearing several different functionalities is one of the major goals of modern synthetic organic chemistry as these structures exist in numerous pharmaceutical and natural products.1 The stereocontrolled formation of such complex molecules with several adjacent stereogenic centers is regarded as a great challenge, because with an increase of the stereocenters the number of possible stereoisomers also increases exponentially. Recently, organocatalytic domino or cascade reactions have emerged as a powerful strategy for providing these complex molecular frameworks by employing simple and readily available precursors in a simple operational procedure.2 The six-membered carbocycles, i.e. cyclohexane derivatives bearing several adjacent stereogenic centers, are common structural features of many valuable natural products and synthetic bioactive compounds, thus leading to the rapid development of the synthetic strategies for obtaining these structures.3
Most of the organocatalytic strategies for the stereoselective synthesis of functionalized cyclohexanes employ aliphatic aldehydes or α,β-unsaturated analogues as one of the substrates, which are activated by chiral amine catalysts via enamine or iminium ion formation.4 The major problem associated with these transformations is the subsequent dehydration after the aldol reaction leading to the loss of two chiral centers. There are only a few reports on the asymmetric synthesis of fully functionalized cyclohexane derivatives. In 2010 Rodriguez et al.5 observed that two molecules of a nitroalkene react in an organocascade asymmetric Michael–Michael–Henry sequence with 1,2-ketoamides to afford fully functionalized cyclohexane derivatives. This process was later on extended by Huang and co-workers6 by using 1,2-ketoesters instead of 1,2-ketoamides in the presence of a copper complex of a chiral diamine. Chen and co-workers also succeeded to create as many as six stereogenic centers on spirocyclic oxindoles in one-pot tandem reactions promoted by a chiral secondary amine and achiral amines.7 Recently, our group has found that one-pot reactions of β-ketoesters with two different electrophiles, i.e. nitroalkenes and enals, were facilitated by an amino-thiourea catalyst and a stoichiometric amount of an achiral base to afford fully functionalized cyclohexane derivatives in high stereoselectivities.8
It is highly desirable to extend the scope of these one-pot reactions beyond enals and nitroalkenes. Owing to the synthetic challenge of the controlled formation of many stereogenic centers and knowing the advantages associated with organocatalytic one-pot cascade reactions as well as the importance of the synthesis of cyclohexane derivatives, we herein disclose unprecedented one-pot organocatalytic Michael–Michael–Knoevenagel-type 1,2-addition reactions involving β-dicarbonyl compounds, nitroalkenes and α,α-dicyanoolefins. Employing sequential organocatalysis by using a low loading of a bifunctional amino-squaramide9 and a catalytic amount of an achiral base virtually enantiopure cyclohexane derivatives bearing five stereogenic centers with two vicinal tetrasubstituted carbons could be obtained (Scheme 1). To achieve this we have used a chiral amino-squaramide (1 mol%) derived from quinine as catalyst to promote the Michael addition of the β-ketoester 1a to β-nitrostyrene (2a) in dichloromethane and after 24 hours the α,α-dicyanoolefin 3a was added followed by the addition of DBU (10 mol%) in dichloromethane.10 Further stirring the reaction for 24 hours afforded the desired cyclohexane 4a in 53% yield with 98% ee and >30:1 dr (Table 1, entry 1). The excellent diastereoselectivity of 4a may be due to a dynamic kinetic resolution of the Michael adduct via base mediated deprotonation of the acidic proton followed by selective protonation (Scheme 1).11 Further optimization of the reaction conditions by screening different bases and solvents showed that with 20 mol% of the guanidine base triazabicyclodecene (TBD) in dichloromethane provides good yields of 71% and excellent stereoselectivity (entry 6). The use of pseudo-enantiomeric catalyst II leads to the opposite enantiomer of the product in 70% yield and 96% ee with excellent diastereoselectivity (entry 11).
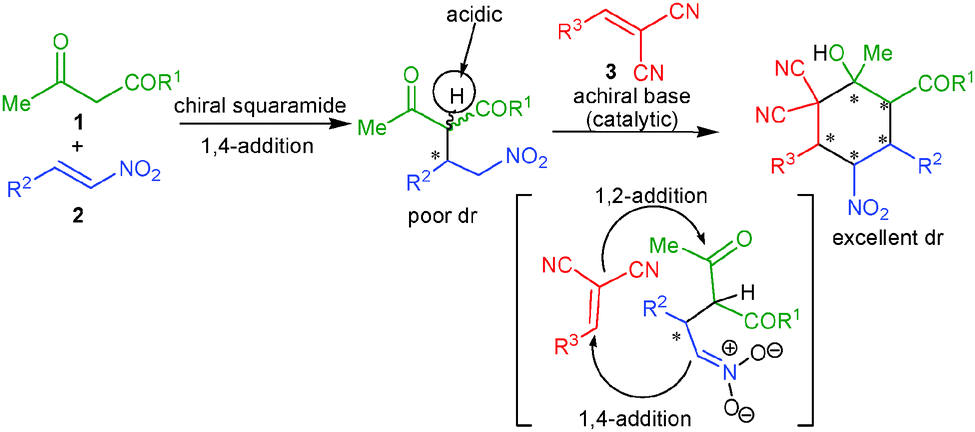 | ||
| Scheme 1 One-pot Michael–Michael–1,2-addition reaction for the asymmetric synthesis of functionalized cyclohexanes bearing five contiguous stereocenters. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Base (x mol%) | Solvent | Yieldb (%) | eec (%) |
|
a Reaction conditions: 0.2 mmol of 1a, 0.2 mmol of 2a, 1 mol% of I, 0.24 mmol of 3a and x mol% of base (0.1 M in solvent).
b Yield of isolated 4a after column chromatography.
c Enantiomeric excess of the major diastereomer (>30:1 dr) determined by HPLC analysis on a chiral stationary phase.
d ee value of ent-4a.
|
||||
| 1 | DBU (10) | CH2Cl2 | 53 | 98 |
| 2 | DBN (10) | CH2Cl2 | 31 | 99 |
| 3 | DABCO (10) | CH2Cl2 | — | — |
| 4 | TEA (10) | CH2Cl2 | 35 | 89 |
| 5 | TBD (10) | CH2Cl2 | 58 | 99 |
| 6 | TBD (20) | CH2Cl2 | 71 | 99 |
| 7 | TBD (30) | CH2Cl2 | 69 | 99 |
| 8 | TBD (20) | CHCl3 | 62 | 99 |
| 9 | TBD (20) | Toluene | 63 | 99 |
| 10 | TBD (20) | THF | 56 | 99 |
| 11 | TBD (20) | CH2Cl2 | 70 | 96d |
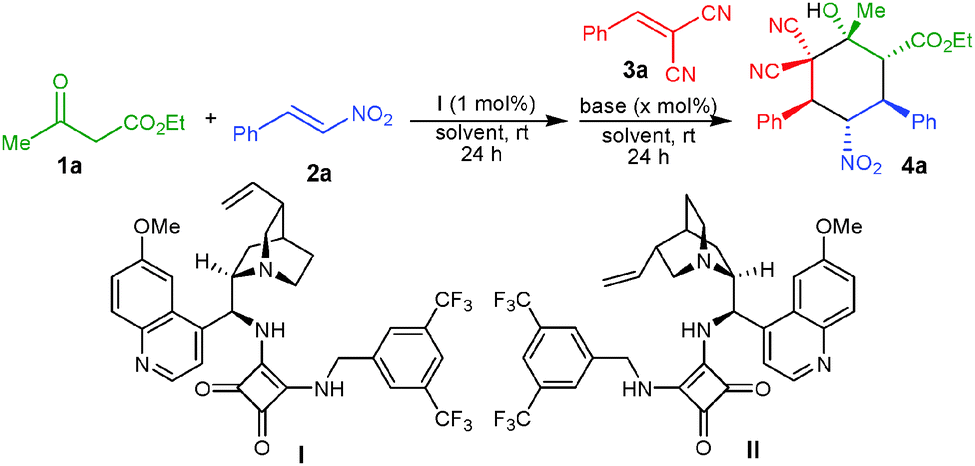
Under optimized reaction conditions, the substrate scope was evaluated at 0.5 mmol scale, which revealed that the use of various α,α-dicyanoolefins bearing electron withdrawing and electron-donating substituents provides a direct access to the corresponding cyclohexanes 4b–g in very good yields (70–84%) and virtually complete enantioselectivity of 99% ee (Table 2, entries 2–7). A α,α-dicyanoolefin with a heteroaromatic group could be employed under the standard reaction conditions, which afforded the desired product 4h in 80% yield and 99% ee (entry 8). However, a α,α-dicyanoolefin bearing a cyclohexyl group did not provide the desired cyclohexane under the optimized reaction conditions. Further screening of different nitroalkenes showed that various electron rich and electron deficient aromatic nitroalkenes as well as heteroaromatic nitroalkenes also worked well under this one-pot procedure to afford fully functionalized cyclohexanes 4i–n in 68–75% and high stereoselectivities (>30:1 dr and 99% ee) (entries 9–14). An aliphatic nitroalkene was also tolerated to afford the corresponding adduct 4o in 69% yield and excellent stereoselectivity (entry 15). Other β-ketoester and a β-diketone were also found to react efficiently to give good yields and excellent stereoselectivities of the corresponding products 4p and 4q (entries 16 and 17).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | 4/ent-4 | Yieldb (%) | eec (%) |
| a Reaction conditions: 0.5 mmol of 1, 0.5 mmol of 2, 1 mol% of I (entries 1–17) or II (entries 18–25), 0.6 mmol of 3 and 20 mol% of TBD (0.1 M in CH2Cl2). b Yield of isolated product after column chromatography. c Enantiomeric excess of the major diastereomer determined by HPLC analysis on a chiral stationary phase. | ||||||
| 1 | OEt | Ph | Ph | 4a | 72 | 99 |
| 2 | OEt | Ph | 4-FC6H4 | 4b | 76 | 99 |
| 3 | OEt | Ph | 4-ClC6H4 | 4c | 72 | 99 |
| 4 | OEt | Ph | 3-ClC6H4 | 4d | 74 | 99 |
| 5 | OEt | Ph | 2-ClC6H4 | 4e | 70 | 99 |
| 6 | OEt | Ph | 4-MeC6H4 | 4f | 84 | 99 |
| 7 | OEt | Ph | 4-MeOC6H4 | 4g | 80 | 99 |
| 8 | OEt | Ph | 2-Thienyl | 4h | 80 | 99 |
| 9 | OEt | 4-FC6H4 | Ph | 4i | 74 | 99 |
| 10 | OEt | 4-ClC6H4 | Ph | 4j | 73 | 99 |
| 11 | OEt | 4-MeC6H4 | Ph | 4k | 68 | 99 |
| 12 | OEt | 3-MeOC6H4 | Ph | 4l | 70 | 98 |
| 13 | OEt | 2-Furanyl | Ph | 4m | 75 | 99 |
| 14 | OEt | 2-Thienyl | Ph | 4n | 68 | 99 |
| 15 | OEt | Cyclohexyl | Ph | 4o | 69 | 99 |
| 16 | OMe | Ph | Ph | 4p | 69 | 99 |
| 17 | Me | Ph | Ph | 4q | 71 | 99 |
| 18 | OEt | Ph | Ph | ent-4a | 70 | 96 |
| 19 | OEt | Ph | 4-FC6H4 | ent-4b | 77 | 97 |
| 20 | OEt | Ph | 3-ClC6H4 | ent-4d | 75 | 98 |
| 21 | OEt | Ph | 2-ClC6H4 | ent-4e | 77 | 98 |
| 22 | OEt | Ph | 4-MeC6H4 | ent-4f | 86 | 96 |
| 23 | OEt | Ph | 2-Thienyl | ent-4h | 81 | 99 |
| 24 | OEt | 4-FC6H4 | Ph | ent-4i | 75 | 98 |
| 25 | OEt | 4-MeC6H4 | Ph | ent-4k | 72 | 99 |
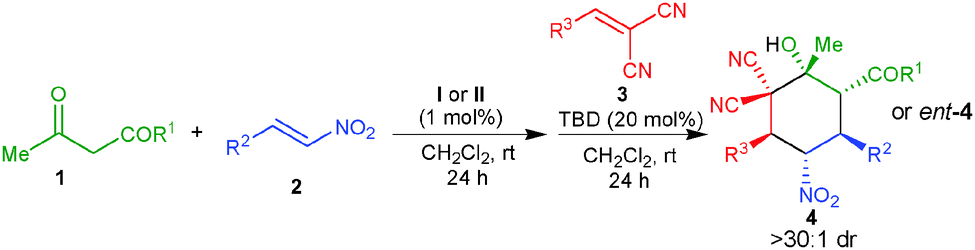
The synthesis of the enantiomers of the products is also possible by employing the pseudo-enantiomeric amino-squaramide catalyst II, which afforded the enantiomers of 4a, 4b, 4d, 4e, 4f, 4h, 4i, and 4k in very good yields (70–86%) and excellent stereoselectivities (>30:1 dr and 96–99% ee, entries 18–25).
The absolute configuration of the products 4a–q synthesized by squaramide I was assigned as 1S,2S,3R,4R and 6S on the basis of a X-ray crystallographic analysis of 4a (Fig. 1).12
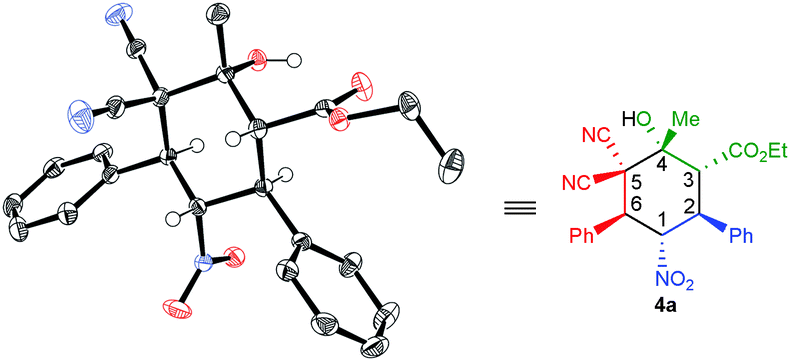 | ||
| Fig. 1 X-ray structure of 4a [Cu-Kα radiation (λ = 1.54178 Å), T = 120 K, Flack parameter: χ = 0.025(115)]. | ||
Further we have tried to extend the substrate scope of this one-pot methodology by employing olefin 5, which gave the adduct 6 bearing six contiguous stereocenters in 67% yield and 99% ee, albeit low diastereoselectivity (Scheme 2).
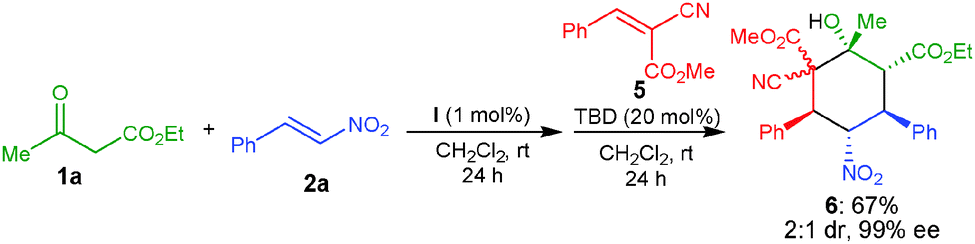 | ||
| Scheme 2 One-pot organocatalytic Michael–Michael–1,2-addition reaction between 1a, 2a and 5. | ||
A one-pot reaction involving the in situ formation of the α,α-dicyanoolefin was successfully performed, which involves the addition of benzaldehyde and malononitrile followed by TBD (40 mol%) to the initially formed Michael adduct of 1a with 2a catalyzed by I (Scheme 3). The corresponding product 4a was obtained in 69% yield, >30:1 dr and 99% ee through this one-pot Michael–Knoevenagel condensation–Michael–1,2-addition sequence.
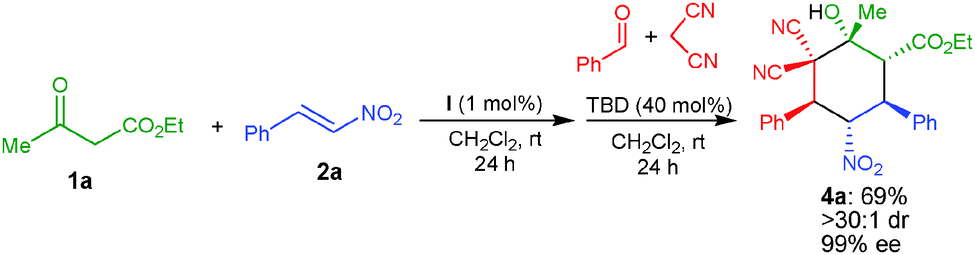 | ||
| Scheme 3 One-pot stereoselective organocatalytic Michael–Knoevenagel condensation–Michael–1,2-addition reaction. | ||
A successful gram-scale reaction between 1a, 2a and 3a to form 4a showed that the reaction efficiency was maintained, thus highlighting the practical and preparative utility of this one-pot process (Scheme 4).
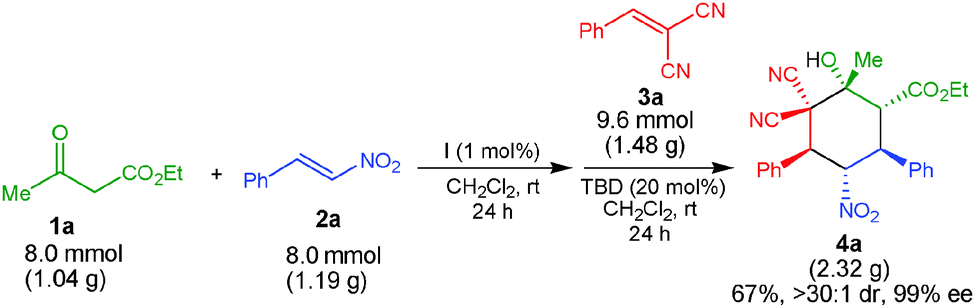 | ||
| Scheme 4 Gram-scale one-pot stereoselective organocatalytic Michael–Michael–1,2-addition reaction. | ||
In conclusion, we have demonstrated the application of a one-pot sequential organocatalysis for the asymmetric synthesis of functionalized cyclohexanes. A low loading of a chiral organocatalyst and a low cost commercially available achiral base afford a series of highly substituted cyclohexane derivatives via one-pot Michael–Michael–1,2-addition reactions in very good yields and excellent stereoselectivities. The enantiomers of the multifunctionalized cyclohexanes are easily accessible by employing a pseudo-enantiomeric amino-squaramide catalyst. This method can be scaled up without any loss of reaction efficiency.
Support from the European Research Council (ERC Advanced Grant “DOMINOCAT”) is gratefully acknowledged.
Notes and references
- For reviews, see: (a) K. C. Nicolaou, T. Montagnon and S. A. Snyder, Chem. Commun., 2003, 551 RSC; (b) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef CAS PubMed.
- For selected reviews on organocatalytic domino–cascade reactions, see: (a) D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., Int. Ed., 2007, 46, 1570 CrossRef CAS PubMed; (b) X. Yu and W. Wang, Org. Biomol. Chem., 2008, 6, 2037 RSC; (c) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed; (d) Ł. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492 CrossRef PubMed; (e) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314 CrossRef CAS PubMed; (f) H. Pellissier, Adv. Synth. Catal., 2012, 354, 237 CrossRef CAS; (g) P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2014, 53, 1485 CrossRef CAS PubMed.
- For reviews on carbocycles, see: (a) C. Cismas, A. Terec, S. Mager and I. Grosu, Curr. Org. Chem., 2005, 9, 1287 CrossRef CAS; (b) J. Wolfling, ARKIVOC, 2007, 5, 210 CrossRef; (c) A. M. Shestopalov, A. A. Shestopalov and L. A. Rodinovskaya, Synthesis, 2008, 1 CrossRef CAS PubMed; (d) J. Shen and C.-H. Tan, Org. Biomol. Chem., 2008, 6, 3229 RSC; (e) S. Reymond and J. Cossy, Chem. Rev., 2008, 108, 5359 CrossRef CAS PubMed; (f) S. Goudedranche, W. Raimondi, X. Bugaut, T. Constantieux, D. Bonne and J. Rodriguez, Synthesis, 2013, 1909 CAS.
- (a) D. Enders, M. R. M. Hüttl, Y. Runsink, G. Raabe and B. Wendt, Angew. Chem., Int. Ed., 2007, 46, 467 CrossRef CAS PubMed; (b) D. Enders, M. R. M. Hüttl, G. Raabe and J. W. Bats, Adv. Synth. Catal., 2008, 350, 267 CrossRef CAS; (c) P. G. McGarraugh and S. E. Brenner, Org. Lett., 2009, 11, 5654 CrossRef CAS PubMed; (d) Y. Wang, R.-G. Han, Y.-L. Zhao, S. Yang, P.-F. Xu and D. J. Dixon, Angew. Chem., Int. Ed., 2009, 48, 9834 CrossRef CAS PubMed; (e) D. Enders, B. Schmid and N. Erdmann, Synthesis, 2010, 2271 CrossRef CAS PubMed; (f) M. Rueping, K. L. Haack, W. Ieawsuwan, H. Sundén, M. Blanco and F. R. Schoepke, Chem. Commun., 2011, 47, 3828 RSC; (g) C. Cassani, X. Tian, E. C. Escudero-Adán and P. Melchiorre, Chem. Commun., 2011, 47, 233 RSC; (h) A. Zea, A.-N. R. Alba, A. Mazzanti, A. Moyano and R. Rios, Org. Biomol. Chem., 2011, 9, 6519 RSC; (i) D. Enders, A. Greb, K. Deckers, P. Selig and C. Merkens, Chem. – Eur. J., 2012, 18, 10226 CrossRef CAS PubMed; (j) X. Zeng, Q. Ni, G. Raabe and D. Enders, Angew. Chem., Int. Ed., 2013, 52, 2977 CrossRef CAS PubMed.
- (a) O. Baslé, W. Raimondi, M. M. Sanchez Duque, D. Bonne, T. Constantieux and J. Rodriguez, Org. Lett., 2010, 12, 5246 CrossRef PubMed; (b) W. Raimondi, M. M. Sanchez Duque, S. Goudedranche, A. Quintard, T. Constantieux, X. Bugaut, D. Bonne and J. Rodriguez, Synthesis, 2013, 1659 CAS.
- D. Shi, Y. Xie, H. Zhou, C. Xia and H. Huang, Angew. Chem., Int. Ed., 2012, 51, 1248 CrossRef CAS PubMed.
- K. Jiang, Z.-J. Jia, S. Chen, L. Wu and Y.-C. Chen, Chem. – Eur. J., 2010, 16, 2852 CrossRef CAS PubMed.
- D. Enders, G. Urbanietz, E. Cassens-Sasse, S. Keeß and G. Raabe, Adv. Synth. Catal., 2012, 354, 1481 CrossRef CAS.
- For reviews on squaramides, see: (a) J. Alemán, A. Parra, H. Jiang and K. A. Jørgensen, Chem. – Eur. J., 2011, 17, 6890 CrossRef PubMed; (b) R. I. Storer, C. Aciro and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330 RSC ; for examples, see: ; (c) J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416 CrossRef CAS PubMed; (d) H. Y. Bae, S. Some, J. S. Oh, Y. S. Lee and C. E. Song, Chem. Commun., 2011, 47, 9621 RSC; (e) Y.-F. Wang, R.-X. Chen, K. Wang, B.-B. Zhang, Z.-B. Lib and D.-Q. Xu, Green Chem., 2012, 14, 893 RSC; (f) C. C. J. Loh, D. Hack and D. Enders, Chem. Commun., 2013, 49, 10230 RSC.
- For the initial optimization, see ESI†.
- For a review on organocatalytic DKR, see: (a) H. Pellissier, Adv. Synth. Catal., 2011, 353, 659 CrossRef CAS ; for recent examples, see: ; (b) T. Cheng, S. Meng and Y. Huang, Org. Lett., 2013, 15, 1958 CrossRef CAS PubMed; (c) M. Bergeron-Brlek, T. Teoh and R. Britton, Org. Lett., 2013, 15, 3554 CrossRef CAS PubMed; (d) Q. Dai, H. Arman and J. C.-G. Zhao, Chem. – Eur. J., 2013, 19, 1666 CrossRef CAS PubMed.
- CCDC 990847 (for 4a).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 990847. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc01885k |
| This journal is © The Royal Society of Chemistry 2014 |