 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective fluorination of alkyl C–H bonds via photocatalysis†
Choon Wee
Kee
ab,
Kek Foo
Chin
a,
Ming Wah
Wong
b and
Choon-Hong
Tan
*a
aSchool of Physical & Mathematical Sciences, Division of Chemistry and Biological Chemistry, Nanyang Technological University, 21 Nanyang Link, Singapore 637371, Singapore. E-mail: choonhong@ntu.edu.sg
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore
First published on 3rd June 2014
Abstract
We report the generation of cationic N-radicals from Selectfluor® via energy transfer with anthraquinone as a photocatalyst for the fluorination of unactivated C–H bonds.
Fluorinated compounds are of paramount importance in medicinal chemistry.1 Access to diverse fluorinated building blocks has the potential to broaden our existing library of fluorinated drugs. Significant progress has been made in the introduction of fluorine into arenes2 and asymmetric fluorination via electrophilic and nucleophilic fluorine sources.2b,3 Recently, a radical-based approach to introduce fluorine into sp3 carbon centres has received increasing attention.4 C–F bond formation through the generation of C-radicals via functionalized substrates was demonstrated by Li5 and Sammis.6 These studies utilized Selectfluor® and N-fluorobenzenesulfonimide (NFSI) as radical fluorine sources, respectively.
Selective fluorination via alkyl C–H functionalization is highly attractive due to the ubiquity of alkyl C–H bonds and avoidance of the need to pre-functionalise substrates.7 Fluorination of aliphatic, allylic, and benzylic sp3 C–H bonds has been demonstrated by Britton,8 Chambers and Sandford,9 Chen,10 Doyle,11 Groves,12 Lectka,13 Inoue14 and Sanford.15
Two strategies for selective functionalization of unactivated C–H bonds have been coined by Baran and co-workers: innate16 and guided17 C–H activation.18 Pertinent to innate C–H functionalization, the literature on radical chemistry indicates that selective abstraction of unactivated C–H bonds could be achieved via the use of electrophilic/nucleophilic radicals.19 In particular, the use of cationic N-radicals as electrophilic radicals to selectively chlorinate or brominate electron rich C–H bonds has been well documented in the literature.20 However, to the best of our knowledge, analogous fluorination reaction which exploits the selectivity of cationic N-radicals to achieve selective fluorination via C–H functionalization has not been developed. Pertinent to the use of N-radicals in C–H functionalization, Li and co-workers reported preliminary results on the guided fluorination of C–H bonds with an amidyl radical recently.5c
The stable and commercially available Selectfluor® is a well-established electrophilic fluorine source21 and is amenable to structural modification.22 Structurally, it possesses a dicationic core and is thus an attractive starting point to generate cationic N-radicals for C–H functionalization (Scheme 1).
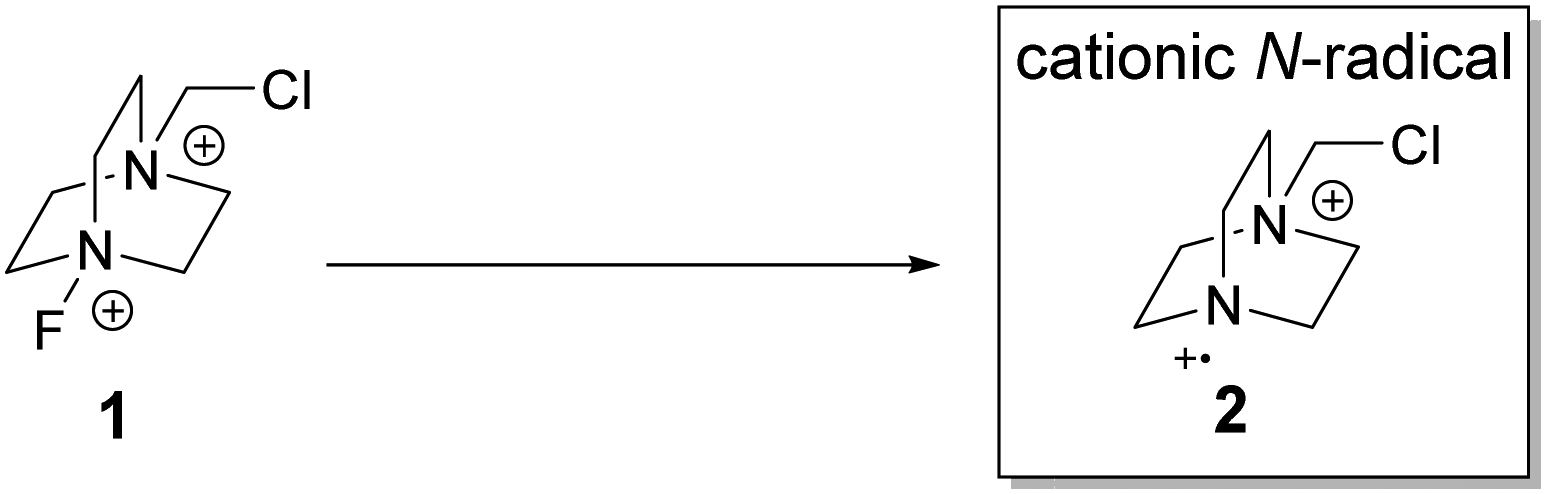 | ||
| Scheme 1 Generation of cationic N-radicals from Selectfluor®. | ||
Photochemistry is an important tool in organic synthesis,23 and in particular, photoredox catalysis has experienced tremendous advancement recently.24 Photochemistry has a long history in C–H functionalization;25 for example, bromination of alkanes with Br2 could be performed with visible light26 and the use of polyoxometalate27 as a photocatalyst in C–H activation.25a Given our interest in C–H functionalization via photochemistry,28 we decided to explore the feasibility of photo-chemically generated cationic N-radicals to selectively fluorinate alkyl C–H bonds.
We found that site selective fluorination of secondary C–H bonds most distal to electron withdrawing groups (EWG) can be achieved using Selectfluor® and a catalytic amount of anthraquinone (AQN). Benzoyl ester 4 was chosen as the model substrate to study the effects of various factors on the fluorination reaction (Scheme 2).
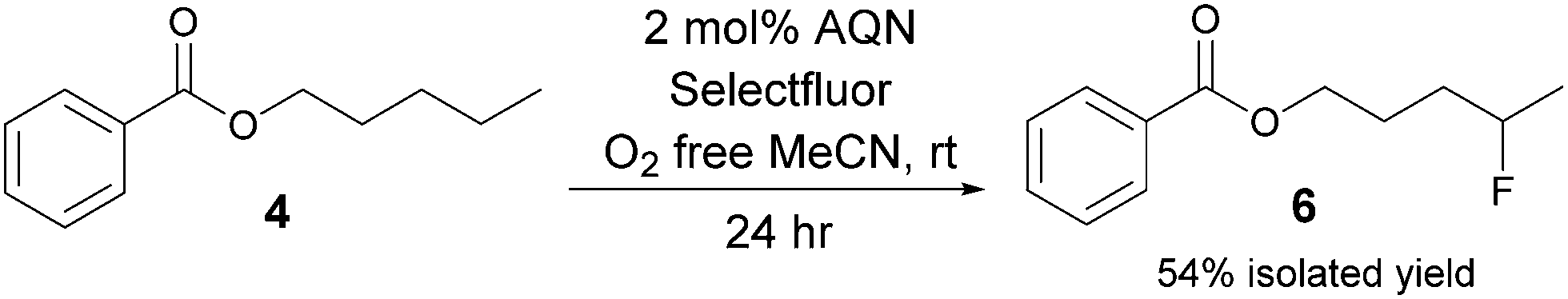 | ||
| Scheme 2 For more details and control experiments please refer to ESI.† | ||
Control experiments established that both AQN and light are essential to the reaction. Triplet dioxygen was found to be detrimental to the reaction (refer to ESI† for more details).
A diverse variety of functional groups can be tolerated in the photo-fluorination (Fig. 1). For aliphatic linear substrates, secondary C–H bonds most distal to the EWG were fluorinated with the highest selectivity. Benzoyl esters of aliphatic alcohols are fluorinated predominantly at the secondary C–H bond most distal to the OBz group (Fig. 1, 5–8). For 8, the tertiary C–H bond is disfavoured due to its proximity to the OBz group; hence selective fluorination of the secondary C–H over the thermodynamically weaker tertiary C–H bond could be achieved. Sulfonate compound 9 gave a similar result to Bz protected compounds.
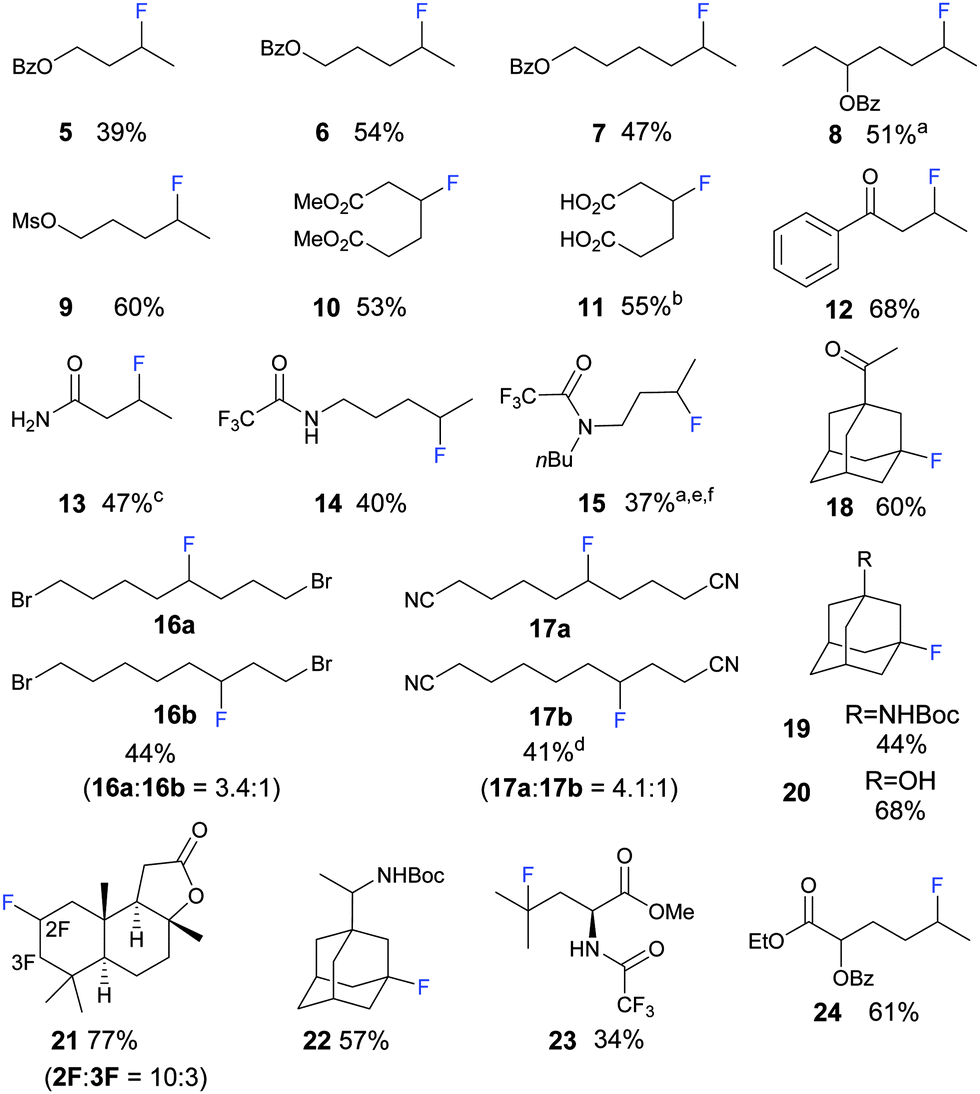 | ||
Fig. 1 Scope of the reaction; protocol: 2 mol% of AQN, substrate: Selectfluor = 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (2 mmol), 8 mL of anhydrous and O2 free MeCN, under Ar and irradiation from an 11 W fluorescent bulb, unless otherwise stated; isolated yield. a Inseparable diastereomers/isomers. b Converted to ester to facilitate separation. c 5 mmol of substrate. d 1.5 equiv. of Selectfluor. e 2 equiv. of substrate. f 2-Cl-AQN (2-chloroanthracene-9,10-dione) was used as a photocatalyst, Rf of AQN and the product is the same on TLC. Only the major product is depicted. For detailed information on the experimental procedure and the ratio of other minor isomeric fluorinated products please refer to ESI.† :1 (2 mmol), 8 mL of anhydrous and O2 free MeCN, under Ar and irradiation from an 11 W fluorescent bulb, unless otherwise stated; isolated yield. a Inseparable diastereomers/isomers. b Converted to ester to facilitate separation. c 5 mmol of substrate. d 1.5 equiv. of Selectfluor. e 2 equiv. of substrate. f 2-Cl-AQN (2-chloroanthracene-9,10-dione) was used as a photocatalyst, Rf of AQN and the product is the same on TLC. Only the major product is depicted. For detailed information on the experimental procedure and the ratio of other minor isomeric fluorinated products please refer to ESI.† | ||
Currently, few methods allow the direct β-functionalization of carbonyl compounds.29 The direct β-fluorination of carbonyl groups such as ester, carboxylic acid, ketone and amide is unknown and can be achieved using this methodology. Methyl esters of adipic acid 10, adipic acid 11 and 1-phenylbutan-1-one 12 were fluorinated at the β-position and were obtained in good yield. For 12, slight dehydrofluorination occurred during flash chromatography, leading to yields lower than expected. Primary, secondary and tertiary amide functional groups are tolerated, although their yields are generally lower. Butyramide could be fluorinated at the β-position on a 5.0 mmol scale; recrystallization yielded 3-fluorobutanamide 13 of high purity. Free amine groups are not tolerated by the photo-fluorination; however, fluorination became viable when the amine is protected with the trifluoroacetyl group. Selectivity of protected amines is similar to that of protected alcohols. Secondary amide of 1-pentylamine was fluorinated at the C–H bond most distal to the amide group. A similar result was observed for tertiary amides of dibutylamine 15. An aldehyde group is not tolerated; an acid fluoride was formed through the fluorination of aldehydes' C–H bond. Alkyl bromides are generally less reactive. The electron withdrawing effect exerted by the bromo group is weaker and thus also resulted in lower selectivity. For example, when 1,8-dibromooctane was used a mixture of 4- and 3-fluorinated compounds was obtained in a ratio of 3.4:1(16a:16b) respectively. Nitriles exhibit similar reactivity and selectivity to the alkyl bromides. Decanedinitrile could be fluorinated to give 5-fluoronitrile 17a and 4-fluoronitrile 17b in a ratio of 4.1:1.
The adamantane core is present in several biologically active molecules such as antiviral drugs and saxagliptin (Type II diabetes therapeutic). Fluorinated methyl ketone adamantane 18, –NBoc amantadine 19, tertiary alcohol adamantane 20, and –NBoc rimantadine 22 were obtained through fluorination at the tertiary position on the admantyl group. Due to the high reactivity of the tertiary C–H bond on the adamantane core,30 some difluorination was observed.
(+)-Sclareolide, a terpenoid from plants with antifungal and cytotoxic properties,31 was subjected to the photo-fluorination. Amongst its 26 sets of C–H bonds, C2 and C3 were selectively fluorinated to give a combined high yield of 77% and a selectivity of 10:3 (21). Protected L-leucine was fluorinated selectively at the tertiary C–H bond furthest from its electron-withdrawing group (23). Fluorination of amino acid via C–H functionalization has also been reported by Britton8 and Inoue.14 Analogous hydroxylation has been achieved by White and co-worker using a Fe catalyst.32 Hydroxyl carboxylic acids (AHAs) are widely used in the cosmetic industry to treat dermatological disorders.33 An ester derivative of 2-hydroxyhexanoic acid, an alpha hydroxyl carboxylic acid, could be fluorinated predictably at the secondary C–H bond most distal from its electron-withdrawing groups (24).
The scalability of the photo-fluorination was tested by fluorinating butyramide on a 25 mmol scale (Scheme 3).
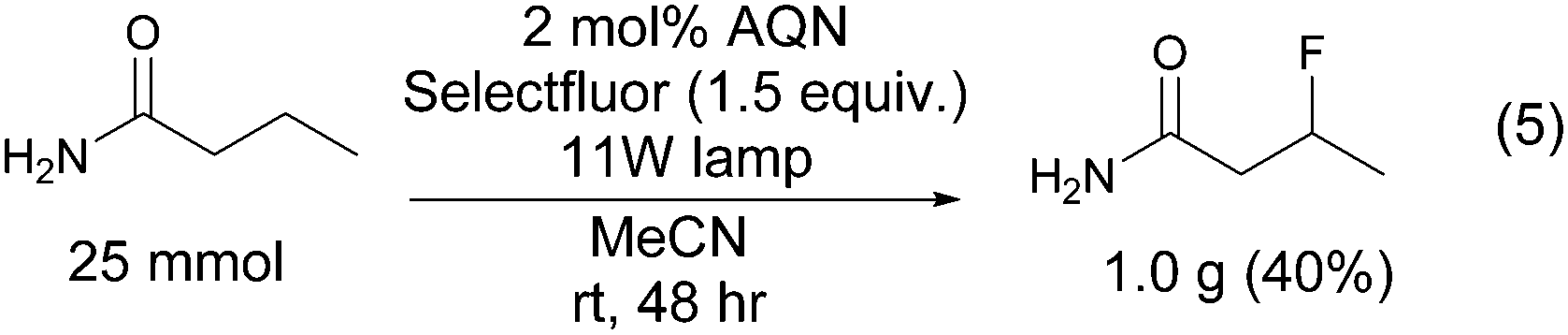 | ||
| Scheme 3 Scaling up. | ||
There are two possible species that can play the role of a hydrogen abstractor: a cationic N-radical and triplet AQN. Triplet benzophenone and its derivatives are well known as hydrogen abstractors,25b while there are less reports on AQN.34 However, triplet benzophenone35 and AQN34a are reported to be nucleophilic radicals. Therefore, they exhibit opposite reactivity observed for the photo-fluorination. Sammis and co-workers have demonstrated that NFSI can be an effective radical fluorine source.6 The insignificant amount of fluorinated products and most importantly, difference in selectivity when Selectfluor® was replaced with NFSI suggest that Selectfluor® is more than a fluorine source.
Hammett analysis36 of the photo-fluorination using AQN gave ρ of −3.1 which correlate with σ+. The ρ of neutral electrophilic hydrogen abstracting radicals are typically from −0.4 to −1.4,37 thus ρ of −3.1 is consistent with the involvement of cationic N-radicals.
Chen and co-workers used fluorenone as the photocatalyst for benzylic fluorination, they proposed that triplet fluorenone is the hydrogen abstractor.10 However, when fluorenone was used to fluorinate 4, an insignificant amount of product was observed. This suggests that a different mechanism is in operation. The reactivity of the photo-fluorination of 4 correlates with the triplet energy (ET) of the photocatalysts, the singlet–triplet gap of 1 (Scheme 4) is 61.4 kcal mol−1, thus triplet–triplet energy transfer39 is feasible between AQN and 1 but not between 1 and fluorenone or alizarin red S salt.
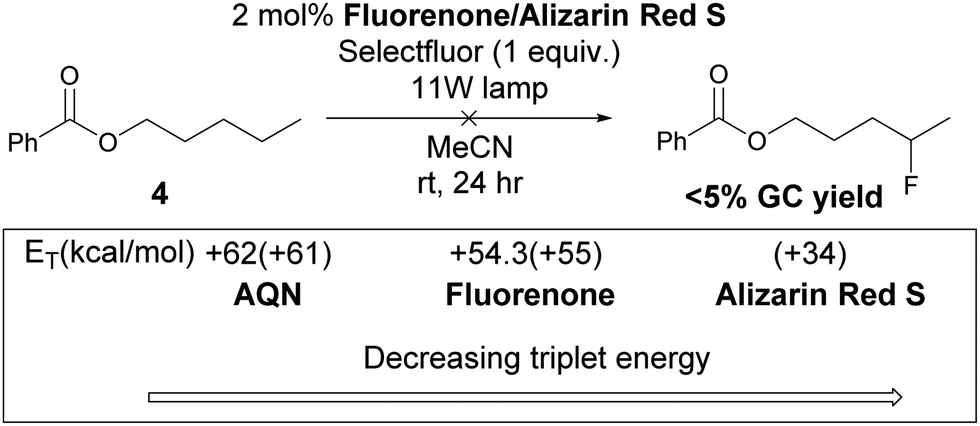 | ||
| Scheme 4 Experiments using relevant photocatalysts. ET of AQN and fluorenone are taken from Zalesskaya.38 Numbers in parentheses are calculated ET. | ||
The selectivity observed for this photo-fluorination resembles that of other reactions using cationic N-radicals.20 Density functional theory40 was used to predict the selectivity of hydrogen abstraction for triplet AQN and cationic N-radicals derived from Selectfluor II® 30. Experimentally, similar selectivity was observed for Selectfluor® and Selectfluor II®. The calculated result shows that 30 has selectivity that is consistent with the experimental results, but not triplet AQN (refer to ESI† for more details).
A preliminary proposal for the mechanism is depicted in Fig. 2. Triplet–triplet energy transfer from 3AQN to 1singlet generates 1triplet. Significant lengthening of the N–F bond was observed when 1singlet is excited to 1triplet. The energy transfer results in the formation of 2 which performs the H abstraction from RH to generate R radicals.
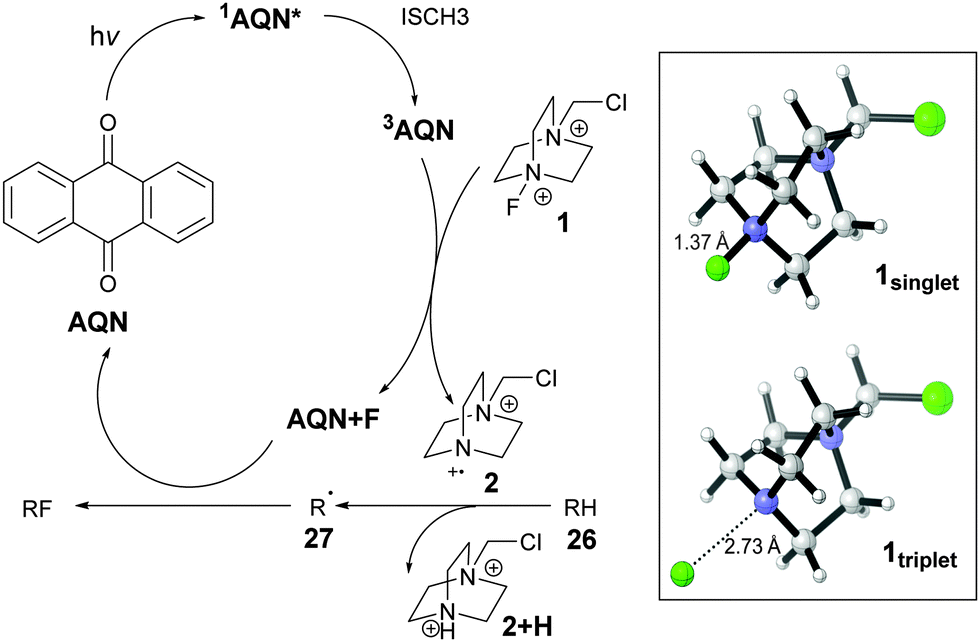 | ||
| Fig. 2 Proposed mechanism. The singlet (1singlet) and triplet (1triplet) state geometries of cations of Selectfluor® are in the box. AQN+F implies a possible complex of fluorine and AQN, as a fluorine radical is not likely to be formed. | ||
In conclusion, we have developed a photo-fluorination reaction. The reaction can be performed using common low power household lamps. The reaction is selective for electron rich sp3 C–H bonds due to the involvement of cationic N-radicals in hydrogen abstraction. This work presents a novel method to generate cationic N-radicals via triplet–triplet energy transfer catalysed by a photocatalyst AQN and extend the scope of innate C–H functionalization of cationic N-radicals to include fluorination. A diverse variety of functional groups can be tolerated by the reaction and it is scalable.
Notes and references
- (a) C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127, 303–319 CrossRef CAS PubMed; (b) K. L. Kirk, Curr. Top. Med. Chem., 2006, 6, 1447–1456 CrossRef CAS; (c) S. Sun and A. Adejare, Curr. Top. Med. Chem., 2006, 6, 1457–1464 CrossRef CAS; (d) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (e) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- (a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed; (b) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- (a) J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1–PR43 CrossRef CAS; (b) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929–2942 RSC.
- M. P. Sibi and Y. Landais, Angew. Chem., Int. Ed., 2013, 52, 3570–3572 CrossRef CAS PubMed.
- (a) Z. Li, C. Zhang, L. Zhu, C. Liu and C. Li, Org. Chem. Front., 2014, 1, 100–104 RSC; (b) C. Zhang, Z. Li, L. Zhu, L. Yu, Z. Wang and C. Li, J. Am. Chem. Soc., 2013, 135, 14082–14085 CrossRef CAS PubMed; (c) Z. Li, L. Song and C. Li, J. Am. Chem. Soc., 2013, 135, 4640–4643 CrossRef CAS PubMed; (d) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401–10404 CrossRef CAS PubMed.
- M. Rueda-Becerril, C. Chatalova Sazepin, J. C. T. Leung, T. Okbinoglu, P. Kennepohl, J.-F. Paquin and G. M. Sammis, J. Am. Chem. Soc., 2012, 134, 4026–4029 CrossRef CAS PubMed.
- (a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed; (b) J.-Q. Yu and Z. Shi, C-H Activation, Springer GmbH, 2010 Search PubMed; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (d) H. M. L. Davies, J. Du Bois and J.-Q. Yu, C-H Functionalization in organic synthesis, The Royal Society of Chemistry, 2011 Search PubMed; (e) M. P. Doyle and K. I. Goldberg, Acc. Chem. Res., 2012, 45, 777 CrossRef CAS PubMed.
- S. D. Halperin, H. Fan, S. Chang, R. E. Martin and R. Britton, Angew. Chem., Int. Ed., 2014, 53, 4690–4693 CrossRef CAS PubMed.
- R. D. Chambers, M. Parsons, G. Sandford and R. Bowden, Chem. Commun., 2000, 959–960 RSC.
- J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494–17500 CrossRef CAS PubMed.
- M.-G. Braun and A. G. Doyle, J. Am. Chem. Soc., 2013, 135, 12990–12993 CrossRef CAS PubMed.
- (a) W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed; (b) W. Liu and J. T. Groves, Angew. Chem., Int. Ed., 2013, 52, 6024–6027 CrossRef CAS PubMed.
- (a) S. Bloom, C. R. Pitts, D. C. Miller, N. Haselton, M. G. Holl, E. Urheim and T. Lectka, Angew. Chem., Int. Ed., 2012, 51, 10580–10583 CrossRef CAS PubMed; (b) S. Bloom, C. R. Pitts, R. Woltornist, A. Griswold, M. G. Holl and T. Lectka, Org. Lett., 2013, 15, 1722–1724 CrossRef CAS PubMed; (c) S. Bloom, J. L. Knippel and T. Lectka, Chem. Sci., 2014, 5, 1175–1178 RSC.
- Y. Amaoka, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 2160–2163 CrossRef CAS PubMed.
- K. B. McMurtrey, J. M. Racowski and M. S. Sanford, Org. Lett., 2012, 14, 4094–4097 CrossRef CAS PubMed.
- S.-Y. Zhang, F.-M. Zhang and Y.-Q. Tu, Chem. Soc. Rev., 2011, 40, 1937–1949 RSC.
- (a) E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70–73 CrossRef CAS PubMed; (b) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed.
- T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826–839 CrossRef PubMed.
- (a) J. M. Tedder, Angew. Chem., Int. Ed., 1982, 21, 401–410 CrossRef; (b) B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC.
- F. Minisci, Synthesis, 1973, 1–24 CrossRef CAS.
- (a) P. T. Nyffeler, S. G. Durón, M. D. Burkart, S. P. Vincent and C.-H. Wong, Angew. Chem., Int. Ed., 2005, 44, 192–212 CrossRef CAS PubMed; (b) S. Stavber and M. Zupan, Acta Chim. Slov., 2005, 52, 13–26 CAS.
- (a) H. P. Shunatona, N. Früh, Y.-M. Wang, V. Rauniyar and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 7724–7727 CrossRef CAS PubMed; (b) J. R. Wolstenhulme, J. Rosenqvist, O. Lozano, J. Ilupeju, N. Wurz, K. M. Engle, G. W. Pidgeon, P. R. Moore, G. Sandford and V. Gouverneur, Angew. Chem., Int. Ed., 2013, 52, 9796–9800 CrossRef CAS PubMed.
- (a) A. G. Griesbeck and J. Mattay, in Synthetic organic photochemistry, Marcel Dekker, New York, 2005, vol. 12, p. 1, online resource (x, 629 p.) ill Search PubMed; (b) T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed; (c) N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed; (d) S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561–574 RSC.
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed; (c) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (d) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC; (e) D. Ravelli and M. Fagnoni, ChemCatChem, 2012, 4, 169–171 CrossRef CAS; (f) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (g) T. P. Yoon, ACS Catal., 2013, 3, 895–902 CrossRef CAS PubMed; (h) J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977–2001 CrossRef PubMed; (i) M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727–2744 CrossRef; (j) D. P. Hari and B. Konig, Chem. Commun., 2014, 50, 6688–6699 RSC.
- (a) C. L. Hill, Synlett, 1995, 127–132 CrossRef CAS PubMed; (b) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS PubMed; (c) I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2011, 50, 1869–1872 CrossRef CAS PubMed.
- G. A. Russell and C. DeBoer, J. Am. Chem. Soc., 1963, 85, 3136–3139 CrossRef CAS.
- M. D. Tzirakis, I. N. Lykakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 38, 2609–2621 RSC.
- C. W. Kee, K. M. Chan, M. W. Wong and C.-H. Tan, Asian J. Org. Chem., 2014, 3, 536–544 CrossRef CAS.
- (a) M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 9886–9887 CrossRef CAS PubMed; (b) A. Renaudat, L. Jean-Gérard, R. Jazzar, C. E. Kefalidis, E. Clot and O. Baudoin, Angew. Chem., Int. Ed., 2010, 49, 7261–7265 CrossRef CAS PubMed; (c) Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984–12986 CrossRef CAS PubMed; (d) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593–1596 CrossRef CAS PubMed; (e) Z. Fu, J. Xu, T. Zhu, W. W. Y. Leong and Y. R. Chi, Nat. Chem., 2013, 5, 835–839 CrossRef CAS PubMed; (f) J. He, M. Wasa, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 3387–3390 CrossRef CAS PubMed.
- T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed.
- R. Atta ur, A. Farooq and M. I. Choudhary, J. Nat. Prod., 1997, 60, 1038–1040 CrossRef PubMed.
- M. S. Chen and M. C. White, Science, 2007, 318, 783–787 CrossRef CAS PubMed.
- S. Kempers, H. I. Katz, R. Wildnauer and B. Green, Cutis, 1998, 61, 347–350 CAS.
- (a) S. Das, J. S. D. Kumar, K. Shivaramayya and M. V. George, J. Chem. Soc., Perkin Trans. 1, 1995, 1797–1799 RSC; (b) N. Tada, K. Hattori, T. Nobuta, T. Miura and A. Itoh, Green Chem., 2011, 13, 1669–1671 RSC.
- S. Kamijo, T. Hoshikawa and M. Inoue, Org. Lett., 2011, 13, 5928–5931 CrossRef CAS PubMed.
- P. R. Wells, Chem. Rev., 1963, 63, 171–219 CrossRef CAS.
- (a) C. Walling and E. A. McElhill, J. Am. Chem. Soc., 1951, 73, 2927–2931 CrossRef CAS; (b) E. S. Huyser, J. Am. Chem. Soc., 1960, 82, 394–396 CrossRef CAS; (c) R. E. Pearson and J. C. Martin, J. Am. Chem. Soc., 1963, 85, 354–355 CrossRef CAS; (d) J. A. Howard, K. U. Ingold and M. Symonds, Can. J. Chem., 1968, 46, 1017–1022 CrossRef CAS.
- G. A. Zalesskaya, J. Appl. Spectrosc., 2002, 69, 328–336 CrossRef CAS.
- (a) Z. Lu and T. P. Yoon, Angew. Chem., Int. Ed., 2012, 51, 10329–10332 CrossRef CAS PubMed; (b) Y.-Q. Zou, S.-W. Duan, X.-G. Meng, X.-Q. Hu, S. Gao, J.-R. Chen and W.-J. Xiao, Tetrahedron, 2012, 68, 6914–6919 CrossRef CAS PubMed; (c) E. P. Farney and T. P. Yoon, Angew. Chem., Int. Ed., 2014, 53, 793–797 CrossRef CAS PubMed.
- M. J. Frisch, et al., in Gaussian 09, Revision A.02, Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc01848f |
| This journal is © The Royal Society of Chemistry 2014 |