 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New ionic dinuclear Ir(III) Schiff base complexes with aggregation-induced phosphorescent emission (AIPE)†
Guangfu
Li
a,
Yong
Wu
a,
Guogang
Shan
a,
Weilong
Che
a,
Dongxia
Zhu
*a,
Baiqiao
Song
a,
Likai
Yan
*a,
Zhongmin
Su
a and
Martin R.
Bryce
*b
aInstitute of Functional Material Chemistry, Faculty of Chemistry, Northeast Normal University, Renmin Road 5268, Changchun, 130024, P.R. China. E-mail: zhudx047@nenu.edu.cn; yanlk924@nenu.edu.cn
bDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 23rd May 2014
Abstract
Two new ionic dinuclear Ir(III) Schiff base complexes which are straightforward to synthesise have luminescence quantum yields as high as 37% in neat films. These are the first examples of dinuclear ionic Ir(III) complexes that display aggregation-induced phosphorescent emission (AIPE).
Solid-state organic luminescent materials are of interest for both fundamental studies and practical applications due to their important roles in such diverse areas as solid-state light-emitting electrochemical cells (LECs),1 chemosensors,2 photocatalysts for hydrogen generation,3 and biochemical probes.4 In this regard understanding and controlling intermolecular interactions is a key topic, as aggregate formation – both in solution and in the solid-state – often quenches light emission.5 In 2001 Tang et al.6 reported an important class of compounds which display aggregation-induced emission (AIE); they emit weakly when dispersed and show strong emission when aggregated or in the solid state. In 2002, Park et al.7 reported aggregation-induced enhanced emission (AIEE) in similar materials. Since then, AIE and AIEE have attracted considerable attention for potential applications in fields such as organic light-emitting devices (OLEDs), bioelectronics and chemosensors.8 It is generally agreed that AIE occurs when molecular packing leads to restricted molecular rotation or torsion which blocks non-radiative channels and effectively suppresses self-quenching. To date only a few neutral iridium complexes with aggregation induced phosphorescent emission (AIPE) have been well studied.9 In 2010, Youngkyu Do et al.10 reported a neutral dinuclear iridium(III) complex which showed AIE character. In 2012, our group11 reported the first ionic mono-nuclear Ir(III) complex which displayed AIE.
However, AIPE materials with high quantum yield are still rare,12 especially ionic transition metal complexes. In this regard, dinuclear Ir(III) complexes are promising candidates. Recently, a series of dinuclear iridium(III) complexes were exploited in LECs.13 It is clear that the nature of the bridging atoms or ligand(s) in dinuclear complexes plays a fundamental role in adjusting the luminescence properties.
Compared with the typical N^N-chelating ligands, such as 2,2′-bipyridine and 1,10-phenanthroline, Schiff base ligands are structurally versatile and can act as both chelating and bridging motifs. The flexible nature of the diimine spacer allows the ligand to bend and rotate freely when coordinating to metal centres so as to adopt the optimum coordination geometries of the metal ions. The motivation for the present work is to study dinuclear iridium complexes with bridging diimine ligands that are able to electronically couple the two metal centres and offer the potential for AIPE.
The complexes 1 and 2 (Scheme 1 and Scheme S1, ESI†) were prepared by the standard procedure14 in high yield. The Schiff-based ligands were synthesised following a previous method (Scheme S1, ESI†).15 The complexes are only very weakly emissive in solution, whereas they are brightly luminescent in solid thin films. Thus, to our knowledge, 1 and 2 present the first examples of ionic dinuclear Ir complexes with AIPE. The crystallographic analysis of the complexes and density functional theory (DFT) gives insight into the origin of their luminescence.
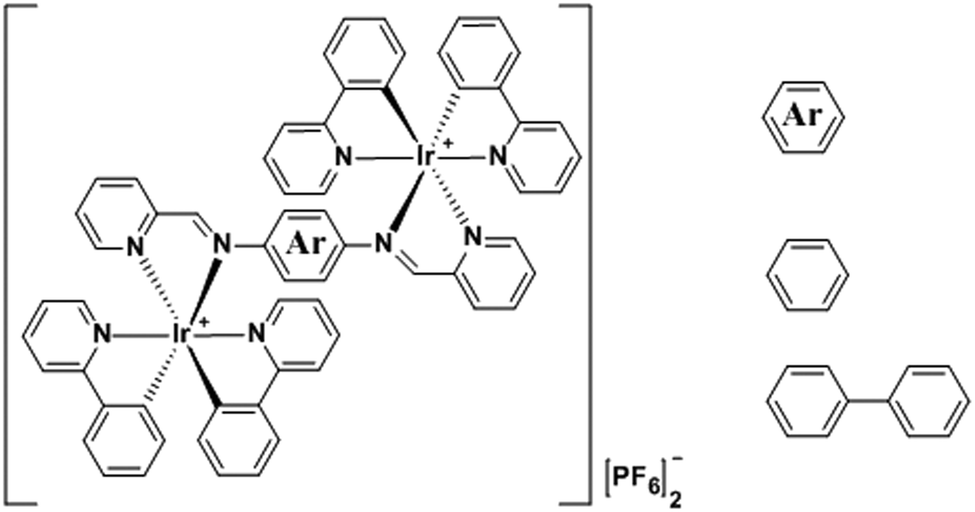 | ||
| Scheme 1 Chemical structures of the complexes, with bridging phenyl (1) and biphenyl (2) units. | ||
Noncovalent intra- and intermolecular effects in single crystals can be used to interpret the origin of the enhanced emission.7,9b The X-ray molecular structure of complex 2 is shown in Fig. 1(a). (For complex 1, see ESI,† Fig. S5). Complex 2 has distorted octahedral geometry around the two Ir centres, which are coordinated by four cyclometalated ligands (C^N, ppy) and one ancillary ligand (N^N), adopting C,C-cis and N,N-trans configurations, as reported for mononuclear ionic iridium complexes.1c,16 The crystal packing of 2 [Fig. 1(b)] shows 1D ladder-like chains, in which the pyridine rings of ppy ligands in adjacent molecules overlap in a face-to-face stack with a separation of ca. 3.667 Å (Fig. S6, ESI†). These intermolecular π–π interactions in 2 restrict the intramolecular motions in the solid state and lead to a more planar geometry within the bridging biphenyl unit. J-aggregation where the molecules are arranged in head-to-head or head-to-tail π–π stacking interactions exists in the crystalline state. DFT calculations show that the π–π interaction between the ppy ligands restricts molecular distortion in the bridging ligand in the solid state (see below). These structural features explain the unusually high solid-state phosphorescence efficiency of 1 and 2.7
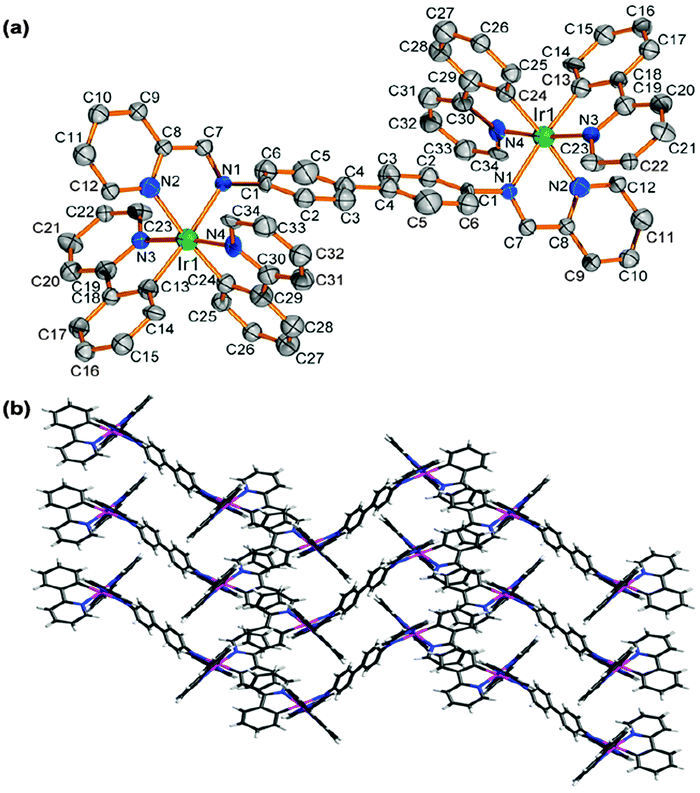 | ||
| Fig. 1 (a) The dication in the X-ray molecular structure of complex 2. The H atoms, PF6− anions and solvent molecules are omitted for clarity. (b) Ladder-like double chain structure formed by π–π stacking in complex 2. | ||
The UV/vis absorption and emission spectra of the complexes 1 and 2 in acetonitrile solution at room temperature are shown in Fig. 2(a) and the data are summarised in Table 1. The dominant absorption band in the 200–300 nm range is assigned to spin-allowed π–π* transitions of the ligands. The weak absorption from 350 nm to the visible region is ascribed to both metal-to-ligand charge transfer (3MLCT) and ligand-to-ligand charge transfer (1LLCT) character with reference to reported Ir(III) complexes.17 Upon photoexcitation, complexes 1 and 2 show almost no emission observable to the naked eye in CH3CN solution [Fig. 2(a)]. In contrast, in neat film the complexes exhibit intense emission with λmax 644 and 692 nm, respectively, at room temperature (Fig. S10, ESI†). This indicates that complexes 1 and 2 may be AIE active. The clear red shift of the crystal emission relative to the powder (Fig. S10 and Table S4, ESI†) can be ascribed to stronger intermolecular interactions in the crystals.18
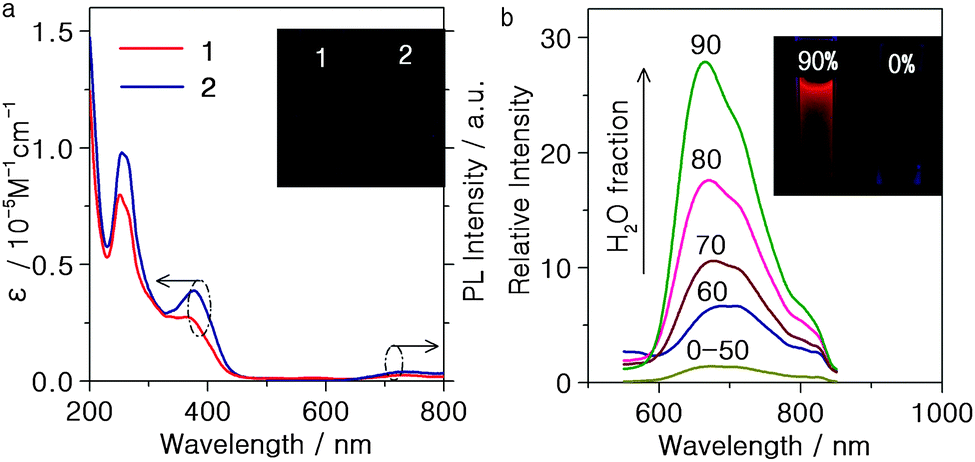 | ||
| Fig. 2 (a) UV-Vis absorption and emission spectra of complexes 1 and 2 (1 × 10−5 M) in CH3CN solution at room temperature. (b) Emission spectra of complex 1 in CH3CN–water mixtures with different water fractions (0–90% v/v) at room temperature. | ||
| λ abs (nm) | λ em (nm) | Φ L | τ (ns) | k r (×106 s−1) | k nr (×107 s−1) | |
|---|---|---|---|---|---|---|
| a Measured in CH3CN (1.0 × 10−5 M) at room temperature. b Measured in the film state at room temperature; λexc = 370 nm; error for ΦL ± 5%. c The radiative kr and non-radiative knr values in neat film were calculated according to the equations: kr = Φ/τ and knr = (1 − Φ)/τ, from the quantum yields Φ and the lifetime τ values. | ||||||
| 1 | 252, 369sh | 644 | 0.373 | 47.66 | 7.83 | 1.32 |
| 2 | 254, 376sh | 692 | 0.264 | 38.68 | 6.83 | 1.90 |
To investigate the AIE properties of 1 and 2, their PL spectra in different ratios of water–acetonitrile mixtures were studied. Both complexes aggregate at high water ratios because 1 and 2 are insoluble in water. As shown in Fig. 2(b), complex 1 in pure acetonitrile exhibits faint emission, but the intensity is remarkably increased when the water fraction exceeds ca. 60%. Complex 2 behaves similarly (Fig. S11(b), ESI†), demonstrating that both 1 and 2 display AIPE. Previous studies have shown that AIE and/or AIEE might be related to the restriction of intermolecular rotation, formation of specific aggregates, and effects of intramolecular planarization of small organic molecules.12 The Schiff base bridging ligands with head-to-head or head-to-tail π–π stacking interactions can provide potential supramolecular recognition sites that can govern the AIPE process.
The absorption spectra of 1 and 2 in different water–acetonitrile ratios are shown in Fig. S13 (ESI†). Both absorption bands of 1 and 2 rise in intensity and move to longer wavelengths when the water fraction increases from 60% to 90%, while the spectra are almost identical in pure acetonitrile solution and 50% water–acetonitrile mixtures. Evidence for aggregates of 1 and 2 in the solvent mixture is seen in the level-off tail in the visible spectral region.6
The PL quantum yields (ΦL) of complexes 1 and 2 in neat film are 37.3% and 26.4%, respectively. Complex 2 is among the most red emissive ionic Ir(III) complex to date19 with its emission maximum red-shifted by 48 nm compared to complex 1. This may be due to the extension of the conjugation length in the ancillary ligand.13a The excited state lifetimes of complexes 1 and 2 in neat film at room temperature (Table 1) are relatively short for phosphorescence materials. Similar values were reported by Youngkyu Do's group.10 The high quantum yields of complexes 1 and 2 can be attributed to their relatively high radiative rates.
To gain further understanding of the unusual solid-state emission properties, the geometries of 1 and 2 were optimised by referring to the X-ray diffraction data, and the electronic properties of the frontier orbitals were studied in solution and the solid state structures using DFT methods. Fig. 3(b) shows that for complex 1 intermolecular CH⋯π interactions among the adjacent ppy ligands exist in the solid state, which induce the intermolecular π–π interactions between the phenyl rings of the cyclometalated ligand and bridge ligand. However, in the solution state for complex 1 there are no π–π interactions so intramolecular relaxation is not restricted. As shown in Fig. 3(a) for complex 2, there is a dihedral angle of 38° between the planes of the two phenyl rings of the bridging ligand in the solution state, whereas in the solid state a planar geometry occurs in the same fragment, probably due to the intermolecular π–π interactions by the adjacent ppy ligands (Fig. 3(b)). For biphenyl and oligo(p-phenylene) derivatives,20 a more planar geometry is favoured in a crystalline environment, even though the distorted geometry is preferred in the isolated state.7 It could, therefore, be concluded that the distorted geometry in solution tends to suppress the radiative process, whereas the planar geometry induced in the solid state activates the radiation.7,21 The HOMO of complex 2 in the solid state (Fig. 4) primarily resides on the two iridium centres and four cyclometalated ppy ligands, whereas in solution only one iridium centre and two cyclometalated ppy ligands participate in the HOMO of 2 (Fig. 4). These results indicate that much more efficient electronic interaction can be induced between the Ir-centred moieties and the bridging ligand in the solid state than in the solution state of 2 due to the planar geometry of the bridge in the solid.10,22 For complex 1 with a phenyl bridge, the HOMO is similar in both the solution and solid states (Fig. 4). The HOMO and LUMO energy levels in solution and solid states are shown in Fig. 4. The energy levels are lowered in the solid state, probably due to intramolecular or intermolecular interactions.
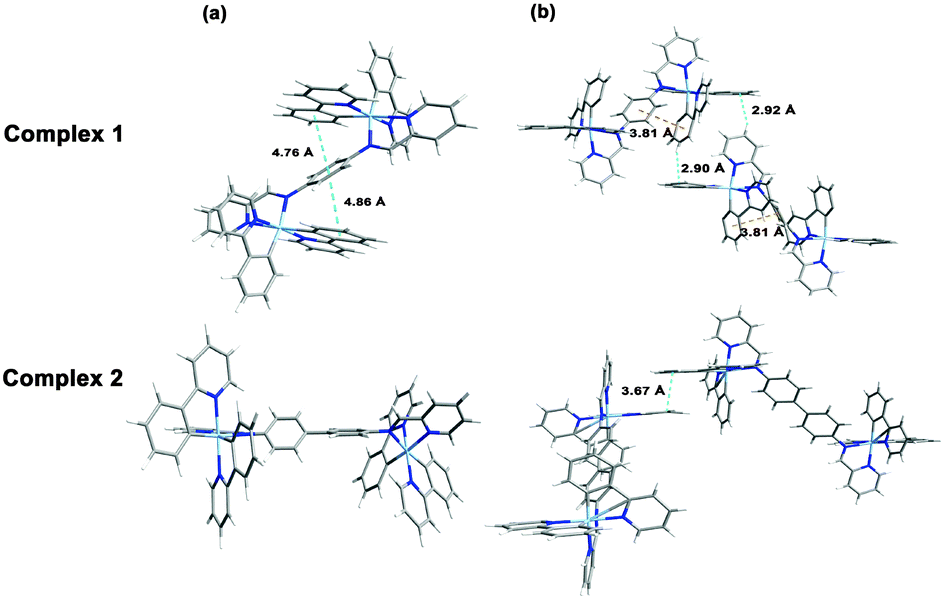 | ||
| Fig. 3 Optimized geometry of complexes 1 and 2 in solution state (a) and the solid state (b). | ||
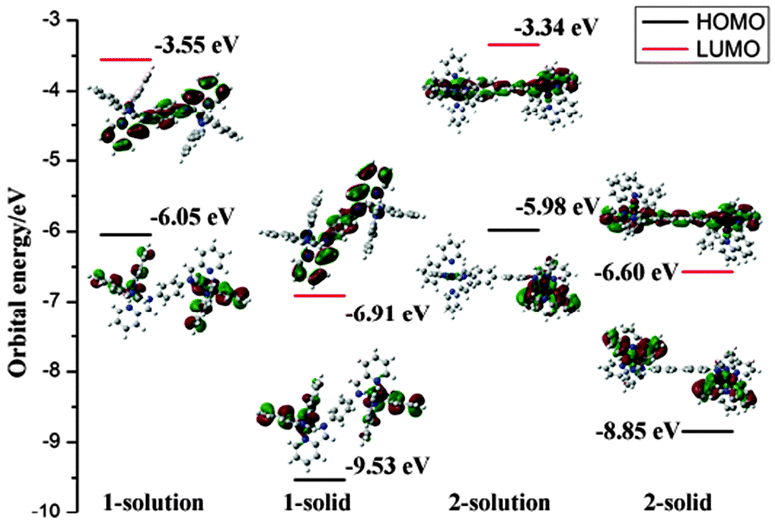 | ||
| Fig. 4 Molecular orbital diagrams, HOMO and LUMO energies for complexes 1 and 2 at their S0 optimized geometries in solution state and solid state. | ||
The emitting triplet states for 1 and 2 are shown to be a mixture of metal-to-ligand charge transfer (3MLCT), ligand-to-ligand charge transfer (3LLCT) and ligand-centered (3LC) transitions (Fig. S15, ESI†). Nonetheless, another important feature should be considered. Table S5 (ESI†) lists the selected calculated bond lengths, bond angles and dihedral angles at both the optimized ground state (S0) and triplet excited state (T1) for 1 and 2. Structural distortions are found in the T1 geometry compared to the S0 geometry in both complexes which induce a larger excited-state relaxation and may result in an effective pathway for nonradiative decay.
In summary, two new ionic dinuclear Ir(III) Schiff base complexes with unusually high PLQY in neat thin films have been studied. X-ray crystal structure analysis and TD-DFT calculations suggest that restricted intramolecular relaxation in the solid state leads to the observed AIPE. This should be a versatile strategy for obtaining highly efficient iTMCs for future applications.
The work in China was funded by NSFC (51203017 and 21303012), the Science and Technology Development Planning of Jilin Province (20100540 and 20130522167JH).
Notes and references
- (a) J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763 CrossRef CAS PubMed; (b) R. D. Costa, E. Ortí, H. J. Bolink, S. Graber, C. E. Housecroft and E. C. Constable, Chem. Commun., 2011, 47, 3207 RSC; (c) R. D. Costa, E. Orti, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178 CrossRef CAS PubMed.
- Q. Zhao, F. Y. Li and C. H. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC.
- A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464 CrossRef CAS PubMed.
- K. K. W. Lo, K. Y. Zhang, S. K. Leung and M. C. Tang, Angew. Chem., Int. Ed., 2008, 47, 2213 CrossRef CAS PubMed.
- H. Rudmann, S. Shimada and M. F. Rubner, J. Am. Chem. Soc., 2002, 124, 4918 CrossRef CAS PubMed.
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS PubMed.
- V. Bhalla, V. Vij, A. Dhir and M. Kumar, Chem. – Eur. J., 2012, 18, 3765 CrossRef CAS PubMed.
- (a) Q. Zhao, L. Li, F. Y. Li, M. X. Yu, Z. P. Liu, T. Yi and C. H. Huang, Chem. Commun., 2008, 685 RSC; (b) Y. You, H. S. Huh, K. S. Kim, S. W. Lee, D. Kim and S. Y. Park, Chem. Commun., 2008, 3998 RSC.
- C. H. Shin, J. O. Huh, S. J. Baek, S. K. Kim, M. H. Lee and Y. K. Do, Eur. J. Inorg. Chem., 2010, 3642 CrossRef CAS.
- G. G. Shan, H. B. Li, J. S. Qin, D. X. Zhu, Y. Liao and Z. M. Su, Dalton Trans., 2012, 41, 9590 RSC.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- (a) F. Lafolet, S. Welter, Z. Popovic and L. De Cola, J. Mater. Chem., 2005, 15, 2820 RSC; (b) C. Sabatini, A. Barbieri, F. Barigelletti, K. J. Arm and J. A. G. Williams, Photochem. Photobiol. Sci., 2007, 6, 397 RSC; (c) C. Ulbricht, B. Beyer, C. Friebe, A. Winter and U. S. Schubert, Adv. Mater., 2009, 21, 4418 CrossRef CAS; (d) R. D. Costa, G. Fernández, L. Sánchez, N. Martín, E. Ortí and H. J. Bolink, Chem. – Eur. J., 2010, 16, 9855 CrossRef CAS PubMed; (e) W. J. Xu, S. J. Liu, X. Zhao, N. Zhao, Z. Q. Liu, H. Xu, H. Liang, Q. Zhao, X. Q. Yu and W. Huang, Chem. – Eur. J., 2013, 19, 621 CrossRef CAS PubMed.
- M. Nonoyama, Bull. Chem. Soc. Jpn., 1974, 47, 767 CrossRef CAS.
- N. M. Shavaleev, Z. R. Bell, G. Accorsi and M. D. Ward, Inorg. Chim. Acta, 2003, 351, 159 CrossRef CAS.
- M. S. Lowry, W. R. Hudson, R. A. Pascal Jr. and S. Bernhard, J. Am. Chem. Soc., 2004, 126, 14129 CrossRef CAS PubMed.
- L. He, J. Qiao, L. Duan, G. Dong, D. Zhang, L. Wang and Y. Qiu, Adv. Funct. Mater., 2009, 19, 2950 CrossRef CAS.
- X. Y. Shen, W. Z. Yuan, Y. Liu, Q. L. Zhao, P. Lu, Y. G. Ma, I. D. Williams, A. J. Qin, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2012, 116, 10541 CAS.
- J. L. Rodriguez-Redondo, R. D. Costa, E. Orti, A. Sastre-Santos, H. J. Bolink and F. Fernandez-Lazaro, Dalton Trans., 2009, 9787 RSC.
- K. N. Baker, A. V. Fratini, T. Resch, H. C. Knachel, W. W. Adams, E. P. Socci and B. L. Farmer, Polymer, 1993, 34, 1571 CrossRef CAS.
- M. M. de Souza, G. Rumbles, I. R. Gould, H. Amer, I. D. W. Samuel, S. C. Moratti and A. B. Holmes, Synth. Met., 2000, 111–112, 539 CrossRef CAS.
- S. Welter, F. Lafolet, E. Ceechetto, F. Vergeer and L. De Cola, ChemPhysChem, 2005, 6, 2417 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental schemes for the complexes, cyclic voltammograms and procedures for the DFT calculations. CCDC 976189 and 970289. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc01799d |
| This journal is © The Royal Society of Chemistry 2014 |