 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical discrimination of phthalic acid among three phthalic acid isomers based on an N-butylaminomethyl-ferrocene derivative†
Jisoo
Yoon‡
a,
Jyoti Ramesh
Jadhav‡
b,
Jun Myung
Kim‡
a,
Minserk
Cheong
a,
Hong-Seok
Kim
*b and
Joohoon
Kim
*a
aDepartment of Chemistry, Research Institute for Basic Sciences, Kyung Hee University, Seoul 130-701, Korea. E-mail: jkim94@khu.ac.kr
bDepartment of Applied Chemistry, Kyungpook National University, Daegu 702-701, Korea. E-mail: kimhs@knu.ac.kr
First published on 27th May 2014
Abstract
A chemosensor compound (1) consisting of a central ferrocene with two butylaminomethyl arms showed unexpected facile electrochemical oxidation of the secondary amines in proximity to the ferrocene, which was utilized for electrochemical discrimination of phthalic acid selectively over two other isomers, isophthalic acid and terephthalic acid.
Dicarboxylic acids play important roles in various biological, industrial, and chemical processes, and have implications in medicine, the environment, and catalysis.1 Considerable efforts have therefore been made in the development of chemical probes that are capable of selectively recognizing and sensing dicarboxylic acids; however, the majority of reported probes are based on NMR and fluorescence studies.2 Recently, electrochemical sensors based on ferrocene, as a redox-responsive unit, have been demonstrated utilizing host–guest complex formation for the selective sensing of specific guest molecules. The ferrocene-based electrochemical sensors employ synthetic receptors that bear several amide, amine, ether, aniline, and triazole moieties for strong and selective recognition of guest molecules mainly involving strong hydrogen-bonding interactions.3 In the electrochemical sensors, the ferrocenes are located in the proximity of the recognition receptor units, exhibiting a significant redox potential shift upon the formation of complexes between sensors and guest molecules. Nevertheless, electrochemical discrimination of isomers has been rarely reported because of very similar chemical and physical properties, which are in general difficult to be electrochemically differentiated, of the isomers. Here, we report a new type of ferrocene-based chemosensor 1 that is capable of discriminating phthalic acid isomers. Compound 1 consisted of a central ferrocene with two simple butylaminomethyl arms (Fig. 1a) and showed unexpected facile electrochemical oxidation of the secondary amines in proximity to the central ferrocene moiety. Interestingly, the facile electrochemical oxidation of the secondary amines in 1 could be utilized to detect the phthalic acid selectively over the two other isomers, isophthalic acid and terephthalic acid. The anodic peak current, which originated from the electrochemical oxidation of the secondary amines in 1, diminished selectively upon the addition of phthalic acid to 1. This enabled selective electrochemical discrimination of the phthalic acid among the three phthalic acid isomers. The phthalic acid-selective electrochemical switch of 1 was also found to be reversible. This electrochemical methodology for discrimination of phthalic acid isomers has been hardly explored, to the best of our knowledge, and provides feasibility to design new electrochemical sensors for unambiguous discrimination of a variety of isomers as arylaminium radical formation has been recently studied for selective sensing of metal ions.4
 | ||
| Fig. 1 (a) Synthetic pathways of 1 and 2. (b) CVs of 1 (solid line) and 2 (dotted line) in DMSO containing 0.1 M TBAP. Concentration: 0.5 mM. Scan rate: 100 mV s−1. | ||
The requisite sensors 1 and 2 were synthesized using the synthetic method depicted in Fig. 1a. For the synthesis of 1, 1,1′-diformylferrocene was converted to its Schiff-base by condensation with 1-butylamine in dry methanol at room temperature, and the latter was reduced with NaBH4 at 0 °C. Compound 2 was also synthesized as a control sensor from 1 with Boc2O in methanol. The structures of sensors 1 and 2 were confirmed by 1H NMR, 13C NMR, and HR-FAB mass analysis (ESI,† see Experimental for details).
The electrochemical behaviors of 1 were investigated by cyclic voltammetry and differential pulse voltammetry in DMSO containing 0.1 M tetrabutylammonium perchlorate (TBAP) as a supporting electrolyte. As shown in Fig. 1b (solid line), we observed an irreversible oxidation peak at 0.02 V (vs. Ag/Ag+) as well as reversible redox peaks centered at 0.30 V (vs. Ag/Ag+) with a peak separation of ca. 85 mV in the cyclic voltammogram (CV) of 1. The reversible redox peaks were attributed to the redox process of the ferrocene moiety of compound 1, but showed a positive potential shift of 0.17 V compared to the redox peaks of authentic ferrocene centered at 0.14 V (vs. Ag/Ag+), as reported previously.3a,b,5 We speculated the first irreversible oxidation peak at 0.02 V (vs. Ag/Ag+) to originate from the oxidation of the secondary amines in proximity to the central ferrocene unit, in light of previous reports regarding the electrochemical oxidation of amine groups followed by irreversible chemical reactions.6 It is worth noting that the oxidation potential of the secondary amines in 1 was unexpectedly lower than the oxidation peaks of the usual secondary amines.6b To confirm the assignment of the irreversible anodic peak of 1, compound 2 (which had a similar structure to 1 but the amines were blocked with Boc protecting groups suppressing the electrochemical oxidation process (Fig. 1a)) was thus synthesized and subjected to electrochemical studies under the same conditions as those of 1. Indeed, the irreversible anodic peak disappeared and only the reversible redox peaks centered at 0.15 V (vs. Ag/Ag+), corresponding to the redox process of ferrocene unit in 2, was observed in CV of 2 (Fig. 1b, dotted line). These results clearly indicate that the irreversible oxidation peak at 0.02 V (vs. Ag/Ag+) observed in the CV of 1 originated from the proton-dependent oxidation of the secondary amines in 1. The unexpected facile electrochemical oxidation of the secondary amines in 1 was believed to be primarily due to the ability of ferrocene to stabilize the aminium radicals, which is consistent with the well-known stabilization of the positive charge on α-carbon atoms by ferrocene.7
Similarly, a differential pulse voltammogram (DPV) of 1 exhibited two successive anodic waves, featuring the sequential oxidations of the secondary amines and ferrocene moieties into the respective amino cationic radicals and ferrocenium ions (Fig. 2a, square). However, the electrochemical behavior of 1 in the presence of phthalic acid was significantly altered. Fig. 2a shows that the first anodic wave at 0.02 V (vs. Ag/Ag+) corresponding to the oxidation of the secondary amines in 1 diminished gradually upon the addition of successive amounts of phthalic acid, while the second wave corresponding to the oxidation of ferrocene unit in 1 showed negligible changes. The first anodic peak currents of 1 completely disappeared after the addition of more than one equivalent of phthalic acid, consistent with the complex formation involving hydrogen bonding interactions of the two secondary amines in 1 with one molar equivalent of the dicarboxylic acid. As expected, we observed no significant changes in DPV of 2 even upon the successive addition of phthalic acids since the amines in 2 were already blocked with Boc groups and thus the corresponding anodic wave was absent in the DPV of 2 (ESI,† Fig. S1). For comparison, we also investigated the electrochemical behaviors of 1 in the presence of two other phthalic acid isomers, isophthalic acid and terephthalic acid, and the results showed different behaviors (Fig. 2b and c). The addition of successive amounts of isophthalic acid and terephthalic acid resulted in no substantial decreases in the first anodic peak current but gradual positive shifts in the peak potential of the first anodic wave corresponding to the oxidation of the secondary amines in 1, reflecting the selective complex formation of 1 with the phthalic acid. We carried out a set of additional experiments to verify the selectivity of 1 to the ortho-isomer. Specifically, DPVs of 1 were obtained with hydroxybenzoic acid isomers under the identical conditions depicted in Fig. 2, and we similarly observed the gradual decreases in the first anodic wave corresponding to the oxidation of the secondary amines in 1 only upon the addition of the ortho-isomer, 2-hydroxybenzoic acid, among the three hydroxybenzoic acid isomers (ESI,† Fig. S2). Computational investigations on the interactions of 1 with the three phthalic acid isomers were also performed to reveal the origin of the selective complex formation of 1 with the ortho-isomer, phthalic acid (ESI,† see Experimental for details). The optimized geometries of 1 with the three isomers indicated hydrogen bonding interactions of 1 with the carboxylic acid moieties of the isomers, but 1 exhibited the strongest hydrogen bonding interactions with the ortho-isomer having the highest ΔHinteraction value (−12.1 kcal mol−1) among the three isomers, which suggests the selective complex formation of 1 with the ortho-isomer, phthalic acid (ESI,† Fig. S3).
 | ||
| Fig. 2 DPVs of 1 with 0.00–4.00 equivalents of (a) phthalic acids, (b) isophthalic acids, and (c) terephthalic acids in DMSO containing 0.1 M TBAP. Pulse amplitude: 0.05 V. (d) Normalized changes in the first anodic peak current (I) of 1, (Io − I)/Io where Io represents the first anodic peak current of free 1, observed in the DPVs of 1 with 0.00–4.00 equivalents of acids. Concentration of 1: 0.5 mM. The error bars represent the standard deviation of the normalized changes from three independent measurements. | ||
The changes in the first anodic peak current (I) of 1 upon the addition of phthalic acid isomers were normalized to the anodic peak current (Io) of free 1, i.e. (Io − I)/Io, and the results, as summarized in Fig. 2d, showed distinctive trends. Fig. 2d clearly shows that the normalized changes, (Io − I)/Io, in the first anodic peak current (I) of 1 increased linearly upon the addition of successive amounts of phthalic acid until the addition of one equivalent of the acid, and then remained constant while the addition of the two other isomers resulted in relatively negligible changes, indicating unambiguous electrochemical discrimination of the phthalic acid among the three different isomers. More significantly, there was an insignificant change in the normalized trend of 1 upon the successive addition of phthalic acid in the presence of two other isomers in excess amounts (four equivalents), suggesting that 1 can be used as a selective electrochemical chemosensor for the phthalic acid even in the presence of other isomers (Fig. 2d, top-down triangle).
Chemosensor 1 also showed reversible transitions between the complexed (OFF) and free (ON) forms, providing reversible electrochemical switches of 1. As already demonstrated above, the anodic wave corresponding to the oxidation of the secondary amines in 1 disappeared (OFF) upon the addition of one and a half equivalents of phthalic acid due to the complete complex formation of 1 (Fig. 3, curve ii). The complexed 1 could then be fully refreshed upon the addition of one and a half equivalents of DBU, a non-nucleophilic base, resulting in the re-appearance (ON) of the anodic wave (Fig. 3, curve iii). The refreshed 1 was exposed to three equivalents of phthalic acid again and showed re-disappearance (OFF) of the anodic wave, indicating re-complexation of 1 with the phthalic acid (Fig. 3, curve iv). These results clearly demonstrate that the phthalic acid-selective electrochemical switch of 1 was reversible, allowing for multiple uses of 1 for the detection of phthalic acid as a molecular switch.
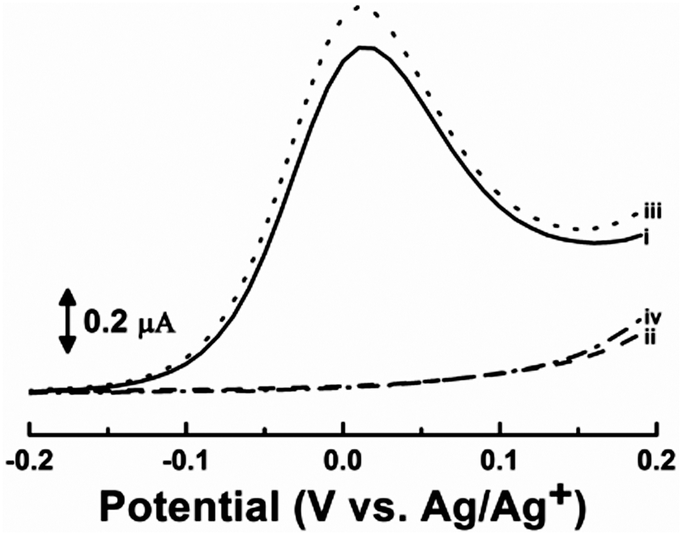 | ||
| Fig. 3 DPVs of (i) free 1, (ii) 1 with 1.50 equivalents of phthalic acid, (iii) 1 with 1.50 equivalents of phthalic acid and DBU, and (iv) 1 with 3.00 equivalents of phthalic acid and 1.50 equivalents of DBU in DMSO containing 0.1 M TBAP. Concentration of 1: 0.5 mM. Pulse amplitude: 0.05 V. | ||
In summary, we reported a ferrocene-based chemosensor 1 exhibiting the electrochemical response turned on with phthalic acid selectively over two other isomers. Compound 1 consisted of a central ferrocene with two simple butylaminomethyl arms, and showed unexpected facile electrochemical oxidation of the secondary amines in proximity to the central ferrocene moiety, attributable to the ability of ferrocene to stabilize the aminium radicals. The addition of phthalic acid to 1 induced a significant decrease in the anodic peak current originating from the electrochemical oxidation of the secondary amines in 1, ascribable to the selective complex formation of 1 with the ortho-isomer, phthalic acid, involving hydrogen bonding interactions of 1 with the carboxylic acid moieties of the phthalic acid. The phthalic acid-selective electrochemical response of 1 was also completely reversible, allowing the repeated uses of 1 for the detection of phthalic acid.
This work was financially supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT and Future Planning (No. 2012R1A1A1014408; J. Kim, 2013R1A1A2006777; H.-S. Kim), the Fundamental R&D Program for Core Technology of Materials of the Ministry of Knowledge Economy of Korea (J. Kim), and the Agency for Defense Development through Chemical and Biological Defense Research Center (J. Kim).
Notes and references
- (a) G. Mingrone, L. Castagneto-Gissey and K. Macé, Br. J. Clin. Pharmacol., 2013, 75, 671 CAS; (b) X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 2010, 385, 1 CrossRef CAS PubMed.
- (a) F. Garcia-Tellado, S. Goswami, S.-K. Chang, S. J. Geib and A. D. Hamilton, J. Am. Chem. Soc., 1990, 112, 7393 CrossRef CAS; (b) S. Goswami, N. K. Das, D. Sen, G. Hazra, J. H. Goh, Y. C. Sing and H.-K. Fun, New J. Chem., 2011, 35, 2811 RSC; (c) S. K. Kim, B.-G. Kang, H. S. Koh, Y. J. Yoon, S. J. Jung, B. Jeong, K.-D. Lee and J. Yoon, Org. Lett., 2004, 6, 4655 CrossRef CAS PubMed.
- (a) D.-S. Kim, H. Miyaji, B.-Y. Chang, S.-M. Park and K. H. Ahn, Chem. Commun., 2006, 3314 RSC; (b) J.-T. Hong, N.-N. Bui, S.-I. Mho, W.-S. Cho and H.-Y. Jang, Bull. Korean Chem. Soc., 2008, 29, 2097 CrossRef CAS; (c) Q.-Y. Cao, T. Pradhan, M. H. Lee, K. No and J. S. Kim, Analyst, 2012, 137, 4454 RSC.
- (a) J. Y. Jung, M. Kang, J. Chun, J. Lee, J. Kim, J. Kim, Y. Kim, S.-J. Kim, C. Lee and J. Yoon, Chem. Commun., 2013, 49, 176 RSC; (b) E. Sanna, L. Martínez, C. Rotger, S. Blasco, J. González, E. García-Espaňa and A. Costa, Org. Lett., 2010, 12, 3840 CrossRef CAS PubMed.
- A. Benito, J. Cano, R. Martínez-Máñez, J. Soto, J. Payá, F. Lloret, M. Julve, J. Faus and M. D. Marcos, Inorg. Chem., 1993, 32, 1197 CrossRef CAS.
- (a) B. Barbier, J. Pinson, G. Desarmot and M. Sanchez, J. Electrochem. Soc., 1990, 137, 1757 CrossRef CAS PubMed; (b) R. S. Deinhammer, M. Ho, J. W. Anderegg and M. D. Porter, Langmuir, 1994, 10, 1306 CrossRef CAS.
- W. E. Britton, R. Kashyap, M. El-Hashash, M. El-Kady and M. Herberhold, Organometallics, 1986, 5, 1029 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and the additional data described in the text. See DOI: 10.1039/c4cc01659a |
| ‡ Equally contributed to this research. |
| This journal is © The Royal Society of Chemistry 2014 |