 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu(I)-mediated 18F-trifluoromethylation of arenes: Rapid synthesis of 18F-labeled trifluoromethyl arenes†
T.
Rühl
a,
W.
Rafique
a,
V. T.
Lien
b and
P. J.
Riss
*ab
aKjemisk Institutt, Universitetet I Oslo, Sem Sælands vei 26, 0371, Oslo, Norway. E-mail: Patrick.Riss@kjemi.uio.no
bNorsk Medisinsk Syklotronsenter AS, Postboks 4950 Nydalen, 0424, Oslo, Norway
First published on 16th April 2014
Abstract
This report is concerned with an efficient, CuI-mediated method for the radiosynthesis of [18F]trifluoromethyl arenes, abundant motifs in small molecule drug candidates and potential radiotracers for positron emission tomography. Three 18F-labelled radiotracer candidates were synthesised from [18F]fluoride ions as proof of principle. The new protocol is widely applicable for the synthesis of novel radiotracers in high radiochemical yields.
Molecular imaging with positron emission tomography (PET) allows for non-invasive, quantitative studies of radiotracer distribution in living subjects. In consequence of its maturation, PET is being increasingly used in routine clinical diagnosis, commercial drug development, and in biomedical research. Novel radiotracers for imaging a variety of biological targets are continually needed to fully exploit the potential of PET.118F is the most frequently employed PET nuclide, due to the extensive use of 2-[18F]fluoro-2-deoxy-D-glucose ([18F]FDG) for clinical diagnosis.2,3 The relevance of 18F is based on its expedient half-life (109.7 min) rendering it suitable for multi-step reactions, transport of radiotracers over moderate distances, convenient handling of the tracer in imaging studies and high-yield cyclotron production of no-carrier-added (n.c.a) [18F]fluoride ions. The ability to form stable C–F bonds promotes the straight introduction of F atoms into most small organic molecules.
Despite the strong demand for novel radiotracers for a variety of disease related biological mechanisms, radiotracer development is a complex process. Researchers and clinicians often struggle to obtain a desired radiotracer within a reasonable time frame because discovery of suitable molecular structures that can be labelled by established procedures often require time-consuming iterative cycles of candidate synthesis and biological evaluation.
Due to its properties, a wide portfolio of synthetic drug molecules and derivatives contain the metabolically stable CF3 group, and consequently an operationally simple and direct arene-trifluoromethylation methodology has become a key focus in current organic chemistry.4 Radiolabelling these CF3 groups is attractive to reposition known drug molecules for PET.5
We, hence, sought an efficient method for producing [18F]trifluoromethyl arenes starting from [18F]fluoride ions within our radiotracer development program. Radiosynthesis of the 18F-labelled aryl trifluoromethane scaffold has been reported, however, mostly through the use of rare and unavailable electrophilic fluorinating agents or harsh conditions.5 A more recent breakthrough employed CuI in combination with aryl iodides.5a We have explored a new route inspired by a recent report on [18F]fluoroform by Vugts et al.6
For successful outcomes, reactions involving [18F]fluoroform require diligent control of the gaseous intermediate, including low temperature distillation and trapping of the product at −80 °C in a secondary reaction vessel. These conditions and technical requirements are limiting factors with respect to the automated synthesis of high activity batches using automated synthesiser systems. Few commercially available systems provide more than one reactor and generally disfavour low temperature processes. We surmised that widespread adoption of trifluoromethylation reactions would strongly benefit from a straightforward nucleophilic one-pot method generally applicable to latest generation synthetic hardware. Such a methodology would, furthermore, feature direct installation of nucleophilic [18F]fluoride ions into candidate radiotracers to avoid loss of radioactivity, conserve specific radioactivity and achieve rapid and operationally simple radiosynthesis.6 To achieve this we focused our efforts on the in situ formation of a suitable 18F-trifluoromethylating reagent from an appropriate precursor and its direct conversion into the title compounds in the same reaction vessel (Scheme 1).
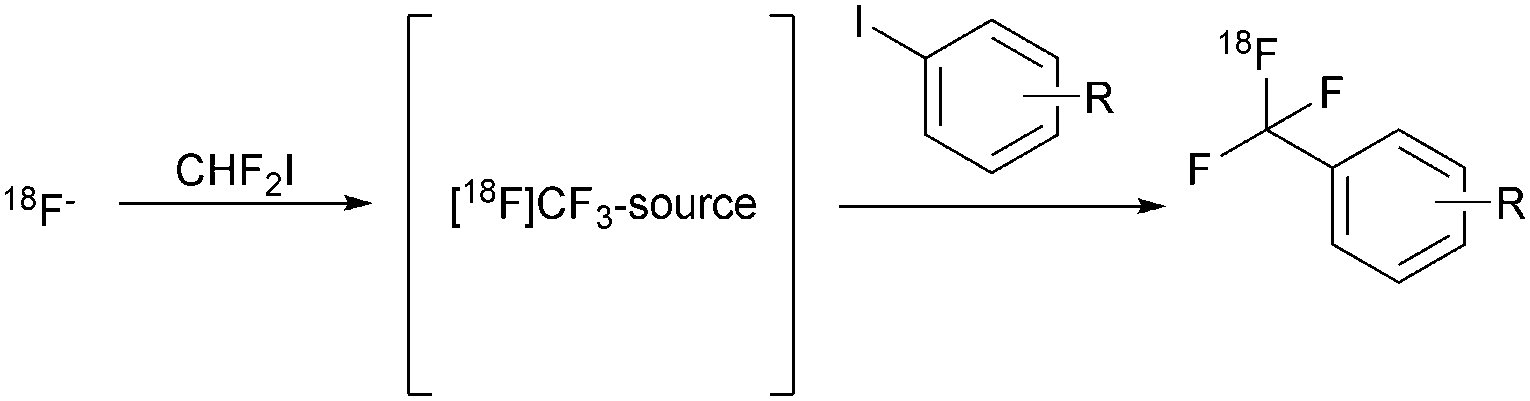 | ||
| Scheme 1 Strategy for the radiosynthesis of [18F]trifluoromethyl arenes. | ||
Difluoroiodomethane (CHF2I) was selected as the starting material to provide a convenient source of Cu[18F]CF3. Our choice of Cu[18F]CF3 was encouraged by the work of Grushin et al.,7a,b who provided comprehensive insights into the formation and use of CuCF3 for trifluoromethylation reactions using fluoroform, which we attempted to implement at first, albeit without success.†
To our dismay, the published reaction conditions translated poorly into radiochemistry.7a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) † This is most likely due to the crucial presence of phase transfer catalysts in the radiofluorination reaction mixture to activate the [18F]fluoride ion. Such reagents are known to increase the basicity and reactivity when combined with strong, anionic bases like KOtBu in dipolar aprotic solvents.7d–f Although such strong bases were deemed crucial to deprotonate fluoroform (pKa = 27 in DMSO) in previous reports,7 only rapid discoloration along with low yields was observed in our radiochemical experiments. Grushin and co-workers described that the use of KOtBu in excess would even permit omission of a supporting ligand and added triethylamine HF-complex to stabilise the Cu–CF3 reagent. Addition of non-radioactive fluoride ions to the labelling reaction is prohibitive in the context of the tracer principle, a prerequisite for PET imaging. A second issue may be the fact that CuF is only stable as a complex in solution and otherwise disproportionates to Cu0 and CuF2. Since the development of a one-pot method would require both species, n.c.a. [18F]fluoride ions and Cu+, to coexist, this mechanism may deprive the reaction mixture of 18F, which is only present in very low concentrations (μM). Hence, obtaining the short-lived [18F]CF3-source in situ prior to trifluoromethylation in the same reaction vessel is rather challenging and, unfortunately, the Grushin method did not furnish the desired labelled products in useful radiochemical yields. Given this apparent incompatibility of reagents a methodological optimisation of various parameters became inevitable.
† This is most likely due to the crucial presence of phase transfer catalysts in the radiofluorination reaction mixture to activate the [18F]fluoride ion. Such reagents are known to increase the basicity and reactivity when combined with strong, anionic bases like KOtBu in dipolar aprotic solvents.7d–f Although such strong bases were deemed crucial to deprotonate fluoroform (pKa = 27 in DMSO) in previous reports,7 only rapid discoloration along with low yields was observed in our radiochemical experiments. Grushin and co-workers described that the use of KOtBu in excess would even permit omission of a supporting ligand and added triethylamine HF-complex to stabilise the Cu–CF3 reagent. Addition of non-radioactive fluoride ions to the labelling reaction is prohibitive in the context of the tracer principle, a prerequisite for PET imaging. A second issue may be the fact that CuF is only stable as a complex in solution and otherwise disproportionates to Cu0 and CuF2. Since the development of a one-pot method would require both species, n.c.a. [18F]fluoride ions and Cu+, to coexist, this mechanism may deprive the reaction mixture of 18F, which is only present in very low concentrations (μM). Hence, obtaining the short-lived [18F]CF3-source in situ prior to trifluoromethylation in the same reaction vessel is rather challenging and, unfortunately, the Grushin method did not furnish the desired labelled products in useful radiochemical yields. Given this apparent incompatibility of reagents a methodological optimisation of various parameters became inevitable.
This prompted us to test our working hypothesis. We surmised that KOtBu may not be required since the formation of difluorocarbene via α-elimination of HI from CHF2I in the presence of 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo [8.8.8]hexacosan (crypt-222), K2CO3, and 18F− facilitates the formation of Cu–[18F]CF3. In addition we considered that a supporting ligand may be beneficial to address the sensitivity of the reaction to Cu-disproportionation and potentially stabilise a Cu–difluorocarbene complex.7g† Consequently, we chose the screening for the most efficient Cu–ligand system in combination with the most frequently used source of reactive 18F-fluoride; crypt-222, K2CO3, and 18F− as the starting point for our investigations, as we now report here.
We initially aimed to discover a simple CuI–ligand system capable of mediating the trifluoromethylation reaction without impeding the nucleophilic radiofluorination with the [18F]fluoride ion. In a model reaction, we chose to use CHF2I, CuI, and a ligand in a 1:1:1 molar ratio (see Table 1) and a model substrate, iodoarene 4-iodo benzonitrile 1a (∼40 μmol) in DMF (0.3 mL). In preliminary experiments (not shown) we found that a temperature of 145 °C was necessary to achieve rapid conversion. Attempts to substitute the low boiling starting material CHF2I (b.p. 22 °C) by a higher boiling difluoromethyl sulfonate, in order to permit better control of the reaction stoichiometry and for easy handling of the reagent, were not successful. Neither difluoromethyl tosylate nor difluoromethyl triflate were found to react to give the desired product under a variety of conditions. Consequently, we resorted to using CHF2I for all further experiments. Despite this minor inconvenience, an activity balance of the reaction did not reveal any loss of the gaseous radioactive material. In control experiments upon omitting either ligand, or CHF2I no 18F-labelled product was obtained (Table 1, entries 1 and 2), likewise, the use of triphenylphosphine gave only traces of the product (Table 1, entry 3). Surprisingly, none of the screened pyridine derivatives gave significant yields of [18F]1b (Table 1, entries 4–7). When the commercially available Cu–NHC complex bis(1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene)copper tetrafluoroborate was used, [18F]1b was formed in about 2% yield (Table 1, entry 8). At this point we deducted that a slightly more basic ligand would be required in dipolar aprotic media and turned our attention to aliphatic, tertiary amines. This hypothesis was rewarded with the first double-figured yield when tetramethylethylenediamine (TMEDA), a ligand that had proved its value previously, was used.8 Under these conditions (Table 1, entry 9) [18F]1b was obtained in 19% radiochemical yield after 10 min at 145 °C. Encouraged by these positive findings we briefly considered DBU (3%, Table 1, entry 10) which turned out to be inferior to TMEDA. Further improved, albeit not yet satisfactory, yield (28%, Table 1, entry 11) was achieved through the use of NEt3 in combination with 1a. Further screening of ligand–catalyst combinations (Table 1, entry 12) revealed N,N-diisopropyl-N-ethylamine (DIPEA) to be very effective with regard to the formation of [18F]1b; without further optimisation a radiochemical yield of 42% was achieved. We hence refrained from further ligand screening and focussed further efforts on the CuI–DIPEA system. Although the majority of [18F]fluoride ions had been consumed within 10 minutes of the reaction time, a considerable amount of residual [18F]fluoride ions left in the reaction mixture indicated further potential for improvement. In order to boost the conversion of [18F]fluoride, which we surmised would lead to further improvements in RCY, we considered that alternative sources of naked [18F]fluoride ions might prove to be beneficial. Sources of the [18F]fluoride ion were obtained by trapping [18F]fluoride ions on a strong anion exchange resin followed by elution of the trapped radioactive material using an appropriate base in aqueous acetonitrile (MeCN–H2O, 9:1). Through this protocol, reactive [18F]fluoride ion complexes are obtained that have found widespread application in PET chemistry. In the context of our one-pot approach we conducted control experiments with DBU and TMEDA alongside DIPEA to avoid overlooking synergies in between the 18F-complex, ligand and CuI. However, under the screened conditions, DIPEA was generally found to be superior to TMEDA and DBU. Substitution of the cryptand crypt-222 (Table 2, entry 1) by the corresponding crown ether 18-crown-6 (Table 2, entry 2) led to a slightly improved radiochemical yield of about 49%. Whereas the use of tetrabutylammonium hydroxide (TBAOH) to form tetrabutylammonium fluoride (TBA[18F]F) (Table 2, entry 3) did not have any effect, the use of tetraethylammonium carbonate (TEAHCO3) to essentially obtain tetraethylammonium fluoride (TEA[18F]F) (Table 2, entry 4) had a remarkable impact (56%). The use of Cs2CO3 as a base, led to a significant increase in the radiochemical yield in the formation of [18F]1b (up to 83%, Table 2, entry 5). In the end, these conditions were equivalent to the combination of KHCO3, crypt-222 and DIPEA which resulted in up to 83% RCY after 10 min at 145 °C (Table 2, entry 6).
| Entry | Liganda | RCYb (%) |
|---|---|---|
| [18F]1b | ||
| a Abbreviations: DBU = 1,8-diazabicyclo[5.4.0]undecene; IPr·CuBF4 = bis(1,3-(2,6-diisopropylphenyl)imidazol-2-ylidene)copper(I) tetrafluoroborate; DMAP = 4-(dimethylamino)pyridine; TMEDA = N,N,N′,N′-tetramethyl ethylenediamine; NEt3 = triethylamine; DIPEA = N,N-diisopropyl-N-ethylamine. b Decay-corrected radiochemical yield in % of dispensed 18F determined by radioHPLC or radioTLC. | ||
| 1 | None | 0 |
| 2 | tBuOK | 2 |
| 3 | Triphenylphosphine | 1 |
| 4 | Pyridine | 2 |
| 5 | DMAP | 9 |
| 6 | 2,2′-Bipyridine | 8 |
| 7 | Phenanthroline | 5 |
| 8 | IPr·CuBF4 | 2 |
| 9 | TMEDA | 19 |
| 10 | DBU | 3 |
| 11 | NEt3 | 28 |
| 12 | DIPEA | 42 |
| Entry | Base | Ligand | [18F]1ba (%) |
|---|---|---|---|
| a Abbreviations: TBAOH = tetrabutylammonium hydroxide; TEAHCO3 = tetraethylammonium bicarbonate. | |||
| 1 | K2CO3/crypt-222 | DIPEA | 42 |
| TMEDA | 19 | ||
| 2 | K2CO3/18-crown-6 | DIPEA | 49 |
| 3 | TBAOH | DIPEA | 2 |
| TMEDA | 18 | ||
| 4 | TEAHCO3 | DIPEA | 56 |
| 5 | Cs2CO3 | DIPEA | 83 |
| TMEDA | 38 | ||
| DBU | 27 | ||
| 6 | KHCO3/crypt-222 | DIPEA | 83 |
| TMEDA | 48 | ||
| DBU | 47 | ||
Notably, screening of various combinations of inorganic bases and phase transfer catalysts used to activate the [18F]fluoride ion in the next step indeed facilitated a duplication of the radiochemical yield even when a milder base was used, highlighting the strong influence of these reagents under our conditions. These conditions were used in all further experiments. In order to further optimise the reaction outcome, we focussed our attention on the contribution of the reaction time. Increasing the reaction time beyond 10 minutes did not improve the yield, instead it became apparent, that the bulk of the [18F]fluoride ions had already been consumed within 10 minutes of the reaction time under our optimised conditions and the yield did not improve, but only degraded further from this point. Substitution of DMF with DMSO or THF was detrimental (Table 3, entries 2 and 4), both of these solvents were ineffective. However, substitution of DMF with MeCN provided a viable alternative and similar yields were obtained. The main disadvantage of using MeCN under these conditions is the fairly pronounced pressure build-up in the reactor, which may result in difficulties during automation. Moreover, a loss of activity was observed using acetonitrile as the solvent.
| Entry | Solvent | Time (minutes) | RCY (%) |
|---|---|---|---|
| 1 | DMF | 10 | 83 |
| 2 | DMSO | 10 | 0 |
| 3 | MeCN | 10 | 78 |
| 4 | THF | 10 | 8 |
| 5 | DMF | 20 | 82 |
Variation of the copper catalyst source. We tested whether CuI was the preferred source of the copper catalyst by changing the copper salt in the promising reaction example that used DIPEA–CuI (Table 4, entry 1). The reaction did not occur when CuI was omitted (Table 4, entry 2). Equimolar replacement of CuI with CuCl, CuOAc, CuCN, or fluorotristriphenylphosphine CuI led to diminished radiochemical yields (Table 4, entries 3–7). Also arene complexes of CuOTf (Table 4, entries 8–10) were not effective in the absence of DIPEA or gave only traces of the 18F-labelled product (Table 4, entry 8). Tetrakis acetonitrile CuOTf and CuBr provided the highest yields (Table 3, entry 4). In our case CuBr was established as the preferred copper source.
| Entry | Catalysta | RCYb (%) |
|---|---|---|
| a Abbreviations: CuOTf·(MeCN)4 = tetrakisacetonitrile copper(I) triflate; CuOTf·benzene = copper(I) trifluoromethane sulfonate benzene complex; CuOTf·toluene = copper(I) trifluoromethane sulfonate toluene complex; CuF·PPh3 = fluorotris(triphenylphosphine)copper(I). b DIPEA was omitted. | ||
| 1 | CuI | 83 |
| 2 | None | 0 |
| 3 | CuCl | 40 |
| 4 | CuBr | 89 |
| 5 | CuCN | 60 |
| 6 | CuOAc | 10 |
| 7 | CuF·PPh3 | 0b |
| 8 | CuOTf·(MeCN)4 | 5b |
| 9 | CuOTf·benzene | 0b |
| 10 | CuOTf·toluene | 0b |
| 11 | CuOTf·(MeCN)4 | 93 |
General conditions, as optimised above, were used to investigate the substrate scope of the 18F-trifluoromethylation. A variety of commercially available aryl halides were screened (Table 5). In essence, aryl iodides were confirmed to be the most appropriate halides for our purpose (Table 5, entry 1), a steep decline in radiochemical yield occurred upon switching to the corresponding bromide 3a or chloride 4a (Table 5, entries 3 and 4). Most assayed functional groups were found to be compatible with the reaction conditions. Potentially sensitive substrates such as 4-cyano or 4-methoxycarbonylbenzenes, which may be sensitive to exposure to carbanionic forms of trifluoromethylating reagents, gave the desired radioactive products in high to excellent yields. Even 12a containing a protic hydroxyl group was tolerated to some extent. The protic carboxamide 10a gave low yield and two unidentified by-products were observed. Electron deficient substrates globally resulted in slightly higher radiochemical yields compared to electron-rich arenes.
|
|
||||
|---|---|---|---|---|
| Substrate | R | X | Product | RCYa,b (%) |
| a Screening conditions: 100 MBq scale, CuBr (58 μmol), CHF2I (169 μmol), KHCO3 (13 μM), crypt-222 (35 μmol), aryl halide (41 μmol), DIPEA (59 μmol), 145 °C, 10 min, DMF (300 μL). b RCY values are mean ± S.D. | ||||
| 1a | 4-Cyano | I | [18F]1b | 93 ± 3 |
| 2a | 4-t-Butyl | I | [18F]2b | 69 ± 8 |
| 3a | 4-t-Butyl | Br | [18F]2b | 1 |
| 4a | 4-t-Butyl | Cl | [18F]2b | 1 |
| 5a | 4-Methoxycarbonyl | I | [18F]5b | 86 ± 7 |
| 6a | 4-Nitro | I | [18F]6b | 89 ± 4 |
| 7a | 4-Pyridinyl | I | [18F]7b | 58 ± 17 |
| 8a | 3-Methoxycarbonyl | I | [18F]8b | 86 ± 8 |
| 9a | 4-Phenyl | I | [18F]9b | 84 ± 2 |
| 10a | 4-Carboxamido | I | [18F]10b | 44 ± 14 |
| 11a | 4-Benzyloxy | I | [18F]11b | 85 ± 6 |
| 12a | 4-Hydroxy | I | [18F]12b | 12 ± 1 |
| 13a | 3,5-Dimethyl | I | [18F]13b | 63 ± 6 |
| 14a | 2,6-Dimethyl | I | [18F]14b | 75 ± 6 |
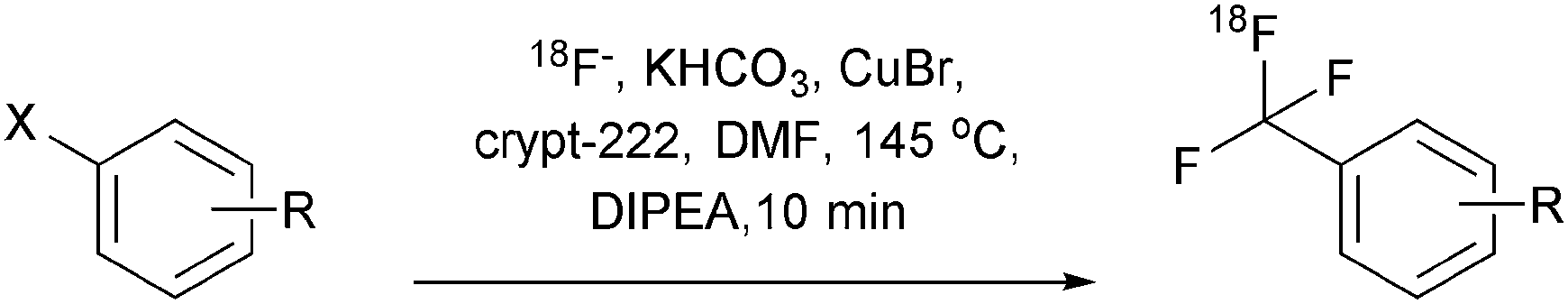
Translation of the method: Radiotracer synthesis. Having confirmed that we were able to prepare a variety of [18F]trifluoromethyl arenes efficiently within only 10 min, we investigated the feasibility of synthesising prospective radiotracer candidates bearing molecular structures common for small molecule drugs (Scheme 2).
 | ||
| Scheme 2 Direct radiosynthesis of [18F]trifluoromethyl arenes. | ||
Treatment of precursor 15a with 18F under our standard conditions afforded the potential subtype selective cannabinoid receptor agonist [18F]15b in 85% RCY. Likewise, we investigated the direct radiosynthesis of trifluorothymine 16b from the corresponding iodide precursor 16a in order to provide this compound for our ongoing cancer imaging efforts in rodent models of peripheral tumours.9 [18F]16b was obtained in a radiochemical yield of 73%. In an extension of our concept the BOC-protected piperazine 17a was converted into the 18F-trifluoromethylated BOC-protected piperazine 17b in 85% yield and deprotected with TFA in a second step to obtain the prospective 5-HT receptor radiotracer 17c.
In this report, we have shown that CuI mediated 18F-trifluoromethylation reactions are highly efficient in the presence of a simple combination of DIPEA, CuBr and iodoarenes. We extended this methodology to three examples of a single-pot synthesis of candidate radioligands for PET imaging (Scheme 2). The resulting [18F]trifluoromethyl arenes were obtained in sufficient yields by using an operationally convenient protocol, suitable for straightforward automation. This direct and rapid conversion of iodoarenes is tolerant to diverse functional groups and consequently provides convenient access to a variety of drug molecules containing the CF3-group. Given the high prevalence of the CF3-group and its prominent role in drug development, paired with the availability of 18F at most PET centres, we expect this novel methodology to be widely adapted for the development of PET radiotracers in particular from known and well characterised drug molecules.
Notes and references
- L. Cai, S. Lu and V. W. Pike, Eur. J. Org. Chem., 2008, 2853–2873 CrossRef CAS.
- (a) P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed; (b) P. J. H. Scott, Angew. Chem., Int. Ed., 2009, 48, 6001–6004 CrossRef CAS PubMed.
- G. J. Meyer, S. L. Waters, H. H. Coenen, A. Luxen, B. Mazière and B. Långström, Eur. J. Nucl. Med., 1995, 22, 1420–1432 CrossRef CAS.
- (a) D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed; (b) A. Deb, S. Manna, A. Modak, T. Patra, S. Maity and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 9747–9750 CrossRef CAS PubMed; (c) S. Mizuta, K. M. Engle, S. Verhoog, O. Galicia-Lopez, M. O'Duill, M. Medebielle, K. Wheelhouse, G. Rassias, A. Thompson and V. Gouverneur, Org. Lett., 2013, 15, 1250–1253 CrossRef CAS PubMed; (d) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed; (e) S. Mizuta, S. Verhoog, K. M. Engle, T. Khotavivattana, M. O'Duill, K. Wheelhouse, G. Rassias, M. Medebielle and V. Gouverneur, J. Am. Chem. Soc., 2013, 135, 2505–2508 CrossRef CAS PubMed.
- (a) M. Huiban, M. Tredwell, S. Mizuta, Z. Wan, X. Zhang, T. L. Collier, V. Gouverneur and J. Passchier, Nat. Chem., 2013, 5, 941–944 CrossRef CAS PubMed; (b) L. Zhu, K. Ploessl and H. F. Kung, Science, 2013, 342, 429–430 CrossRef CAS PubMed; (c) S. Mizuta, I. Stenhagen, M. O'Duill, J. Wolstenhulme, A. Kirjavainen, S. Forsback, M. Tredwell, G. Sandford, P. Moore, M. Huiban, S. Luthra, J. Passchier, O. Solin and V. Gouverneur, Org. Lett., 2013, 15, 2648–2651 CrossRef CAS PubMed; (d) M. Tredwell and V. Gouverneur, Angew. Chem., Int. Ed., 2012, 51, 11426–11437 CrossRef CAS PubMed; (e) P. J. Riss, V. Ferrari, L. Brichard, P. Burke, R. Smith and F. I. Aigbirhio, Org. Biomol. Chem., 2012, 10, 6980–6986 RSC; (f) P. J. Riss and F. I. Aigbirhio, Chem. Commun., 2011, 47, 11873–11875 RSC; (g) M. R. Kilbourn, M. R. Pavia and V. E. Gregor, Int. J. Radiat. Appl. Instrum., Part A, 1990, 41, 823–828 CrossRef CAS; (h) O. Josse, D. Labar, B. Georges, V. Grégoire and J. Marchand-Brynaert, Bioorg. Med. Chem., 2001, 9, 665–675 CrossRef CAS; (i) W. R. Dolbier Jr, A.-R. Li, C. J. Koch, C.-Y. Shiue and A. V. Kachur, Appl. Radiat. Isot., 2001, 54, 73–80 CrossRef CAS.
- D. van der Born, J. D. M. Herscheid, R. V. A. Orru and D. J. Vugts, Chem. Commun., 2013, 49, 4018–4020 RSC.
- (a) A. Lishchynskyi, M. A. Novikov, E. Martin, E. C. Escudero-Adán, P. Novák and V. V. Grushin, J. Org. Chem., 2013, 78, 11126–11146 CrossRef CAS PubMed; (b) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (c) E. A. Symons and M. J. Clermont, J. Am. Chem. Soc., 1981, 103, 3127–3130 CrossRef CAS; (d) M. Halpern, Phase-Transfer Catalysis, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2002 Search PubMed; (e) E. V. Dehmlow and S. S. Dehmlow, Phase Transfer Catalysis, VCH, Weinheim, 3rd edn, 1993 Search PubMed; (f) C. M. Starks, C. L. Liotta and M. E. Halpern, Phase-Transfer Catalysis, Chapman & Hall, New York, 1994 Search PubMed; (g) W. Kirmse, Carbene Chemistry, Academic Press, Inc. LTD, New York-London, 2nd edn, 1971, vol. 1 Search PubMed.
- P. J. Riss, S. Lu, S. Telu, F. I. Aigbirhio and V. W. Pike, Angew. Chem., Int. Ed., 2012, 51, 2698–2702 CrossRef CAS PubMed.
- K. Virdee, P. Cumming, D. Caprioli, B. Jupp, A. Rominger, F. I. Aigbirhio, T. D. Fryer, P. J. Riss and J. W. Dalley, Neurosci. Biobehav. Rev., 2012, 36, 1188–1216 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and analytical data. See DOI: 10.1039/c4cc01641f |
| This journal is © The Royal Society of Chemistry 2014 |