 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioorthogonal site-specific labeling of the 5′-cap structure in eukaryotic mRNAs†
Josephin Marie
Holstein
,
Daniela
Schulz
and
Andrea
Rentmeister
*
Institute of Biochemistry, University of Muenster, Wilhelm-Klemm-Str. 2, D-48149 Münster, Germany. E-mail: a.rentmeister@uni-muenster.de
First published on 12th March 2014
Abstract
We present a chemo-enzymatic approach for site-specific labeling of 5′-capped RNAs based on bioorthogonal chemistry. A trimethylguanosine synthase was engineered to transfer a terminal azido moiety to the 5′-cap which could be further modified using strain-promoted azide–alkyne cycloaddition (SPAAC).
Labeling RNA at specific sites is in most cases a prerequisite to study folding, structural changes and trafficking at the single molecule level or in biological systems. Some complex phenomena like the trafficking of mRNA which leads to subcellular mRNA localization should ideally be studied in living cells. This would require delivery of fluorescently labeled RNA into the cell without affecting the cellular processes, a problem that has not been fully solved. Therefore, most approaches for RNA imaging in living cells extend the RNA of interest by motifs that are recognized and bound by RNA-binding proteins.1 Fluorescent labeling is achieved by fluorescent proteins fused to the RNA-binding proteins. This approach was successfully used to study mRNA trafficking, e.g. ASH1 mRNA in yeast or ubi1 mRNA in the hyphae of fungi.2,3 However, it is at least arguable whether the resulting fluorescing constructs will still show the same behavior as the endogenous RNA, if the RNA to be studied has been extended by multiple RNA-binding motifs (24 can be sufficient for detection4 but up to 96 have been reported5) and multiple fusion proteins of high molecular weight are bound. After all, the appendage will typically exceed the size of the original RNA by far.
To study the target-RNA in the most native form possible, it would be desirable to use small organic dyes instead of extended, non-native sequences plus attached fusion proteins as reporters.
Various methods for the attachment of labels to RNA in vitro or even in cellular systems have therefore been developed. Particularly, approaches based on chemical synthesis and metabolic labeling yielded numerous improvements during the last few years. Metabolic labeling using cell permeable precursors has been achieved with 5-ethynyluridine6 as well as N6-propargyl adenosine.7 The modified nucleotides can be incorporated into RNA at random positions during enzymatic RNA synthesis. An azide-modified UTP analog has been used for transcriptions in vitro.8 In all cases, the introduced terminal alkyne or azido moieties serve as reporter groups for further post-synthetic modification using copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) or Staudinger ligation.
Alternatively, native nucleic acids can be modified site-specifically by enzymatic transfer of reporter groups that are substrates for subsequent chemical modification. Methyltransferases have proven to be particularly useful for this chemo-enzymatic approach. Instead of transferring a methyl group from their natural cosubstrate S-adenosyl-L-methionine (AdoMet, SAM), some methyltransferases show promiscuous activity in cosubstrate analogs and can thus transfer alkene or alkyne groups. Using this approach, tRNA labeling as well as sequence-specific labeling of in vitro produced pre-mRNA was achieved.9,10 The respective RNAs could be further modified using CuAAC.9,10
Regarding studies on RNA trafficking and transport, eukaryotic mRNAs are currently the most interesting study objects within the plethora of RNA species. Not only are they generally transported from the nucleus to the cytoplasm, but certain mRNAs (up to 70% in Drosophila11) can also be actively transported to distinct subcellular regions. Recent findings, suggesting that localized RNAs can be associated with local translation, have attracted considerable attention, particularly for neurons, where this mechanism could be associated with synaptic plasticity or regeneration of axons.3,12–14
An approach that (i) allows attachment of small fluorescent reporters specifically to mRNA, (ii) is compatible with cellular components and (iii) not harmful to living cells would be a significant advance, regarding studies on mRNA trafficking and localization. We recently developed a chemo-enzymatic strategy to specifically label RNAs bearing a 5′-cap—one of the hallmarks of eukaryotic mRNAs.15 Herein, the cap is recognized by an engineered variant of a trimethylguanosine synthase from Giardia lamblia (GlaTgs-Var1) and modified with bioorthogonal groups, namely a terminal alkene or alkyne functionality using analogs of SAM as cosubstrates. The site-specifically introduced alkene or alkyne groups, respectively, were further covalently linked to labels (e.g. fluorescent dye or biotin) using thiol–ene click or CuAAC reactions. The latter could also be carried out in lysates of E. coli and human PC3 cells.
However, although this approach allowed for the first time to label capped RNAs site-specifically, it is not compatible with living cells because the copper concentrations required are toxic.16
To address this issue, we searched for truly bioorthogonal reactions that have been developed to work with living cells or even organisms.
The prime example of a truly bioorthogonal reaction is the strain-promoted azide–alkyne cycloaddition (SPAAC), a reaction between the same functional groups as in CuAAC. However, instead of the copper catalyst, SPAAC benefits from a spring-loaded cyclooctyne. The ring strain of the cyclooctyne greatly facilitates the reaction and makes it work under ambient conditions in the absence of copper. Many differently substituted cyclooctynes have been developed and some of them are now commercially available.17–20 SPAAC has been used to label glycans on the surface of living cells within model organisms like zebrafish,21,22Caenorhabditis elegans22 and mice23 without observable cytotoxicity. Reports on SPAAC for RNA are still scarce and mostly limited to solid phase syntheses.24,25 In combination with enzymes, only the direct incorporation of azide-derivatized nucleotides but not the modification of native RNAs has been reported.26
Our goal was to develop an approach that allows bioorthogonal labeling of native mRNA molecules. Chemo-enzymatic modification could provide a tailor-made strategy for labeling native mRNAs. The native target molecule could be functionalized in a first step by an enzyme capable of transferring a reactive group instead of a methyl group using appropriate SAM analogs as cosubstrates. The specificity of the enzyme should allow us to target a specific subset of RNAs even in the complex cellular environment and even a specific site within the molecule. In a second step, the introduced functionality can be further converted using bioorthogonal click reactions. As a result, a covalent linkage is formed between the target and the reporter molecule. To selectively label mRNAs, we used the enzyme Tgs2 from Giardia lamblia (GlaTgs) that recognizes and normally hypermethylates the 5′-cap—a hallmark of mRNAs—at the exocyclic N2 atom using the cosubstrate SAM. To establish a bioorthogonal strategy, a reactive residue compatible with SPAAC had to be transferred instead of an alkene or alkyne. Thus, either a cyclooctyne or an azido moiety had to be attached. Since this first step is catalyzed enzymatically, we thought it might be unlikely to transfer a sterically demanding cyclooctyne instead of a methyl group and chose to synthesize an azide-bearing SAM analog, 4-azidobut-2-enyl SAM (“Ab-SAM”) 2, which had been successfully used with engineered protein methyltransferases.27 In addition to the terminal azido group, this analog contains an allylic system in the β-position to the sulfonium center, which might stabilize the transition state of the transfer reaction.28
Overall, our approach harnesses GlaTgs2-Var1 (containing the substitution V34A) to enzymatically transfer a terminal azido group to the 5′-cap typical of eukaryotic mRNAs. The terminal azide is then further modified using a fluorescently labeled cyclooctyne for SPAAC (Fig. 1).
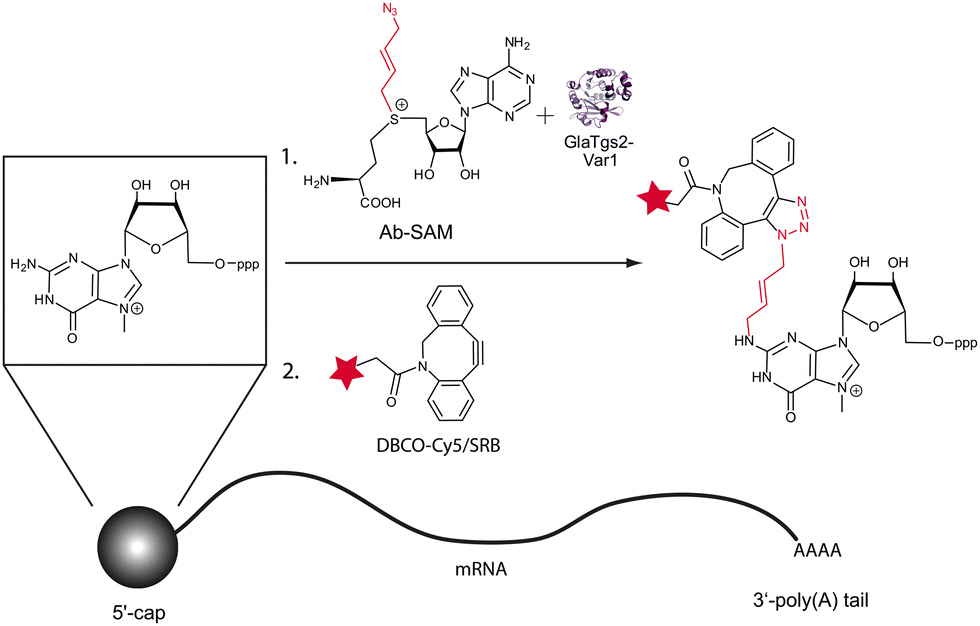 | ||
| Fig. 1 Chemo-enzymatic modification of 5′-capped RNAs using strain-promoted azide–alkyne cycloaddition (SPAAC). A trimethylguanosine synthase variant is able to use the SAM analog “Ab-SAM” as a cosubstrate and transfers the 4-azidobut-2-enyl group to the 5′-cap. The azido functionality reacts with a Cy5- or Sulforhodamine B (SRB)-labeled dibenzylcyclooctyne (DBCO) reagent to form a stable triazol. | ||
We tested the enzymatic conversion of m7GpppA 1, a minimal capped RNA, with Ab-SAM 2 and analyzed the reaction by HPLC and MALDI-TOF-MS (Fig. 2). HPLC analysis showed a new peak at 12 min in reactions containing GlaTgs2-Var1, Ab-SAM 2, and m7GpppA 1. Control reactions performed without enzyme or reactions at time point zero did not show product formation.
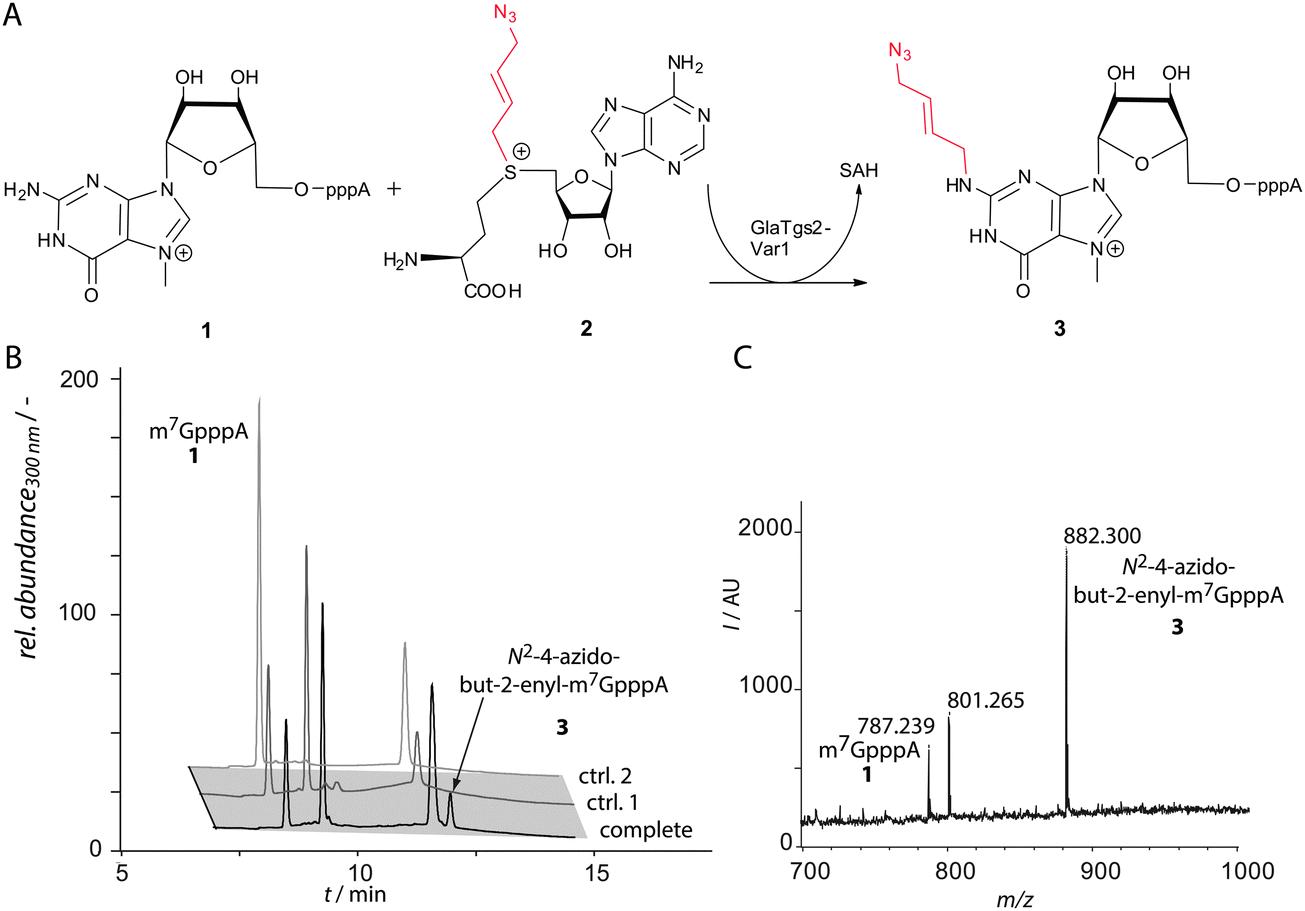 | ||
| Fig. 2 Enzymatic conversion of the cap analog m7GpppA 1 by GlaTgs2-Var1 using Ab-SAM 2 as a cosubstrate. (A) Reaction scheme. GlaTgs2-Var1 generates N2-4-azidobut-2-enyl-m7GpppA 3 using Ab-SAM 2 as a cosubstrate through formation of S-adenosyl-L-homocysteine (SAH). (B) Reversed-phase HPLC analysis of reactions with GlaTgs2-Var1 (90 μM) producing 83 μM 3 (tR = 12 min) in three hours (black). Controls show GlaTgs2-Var1 catalyzed reactions at time point 0 (ctrl. 1) and reactions performed without enzyme (ctrl. 2). (C) MALDI-TOF-MS analysis of the transfer reaction showing m7GpppA 1 (m/z 787) and the formation of N2-4-azidobut-2-enyl-m7GpppA 3 (m/z 882) after three hours. | ||
The product with the correct mass (m/z 882) was found by MALDI-TOF-MS analysis and assigned to 3. Using concentrations of 90 μM GlaTgs-Var1, typically 30% of 1 were converted. As expected, the variant exhibits a markedly lower total turnover number (TTN) for Ab-SAM 2 compared to the natural substrate SAM (TTN 0.8 ± 0.1 vs. 108 ± 6). This can be attributed to the sterically demanding side-chain.
The introduced azido group allows further modifications using SPAAC.29 We tested the commercially available dibenzylcyclooctyne reagents DBCO-Cy5™646/661 4 and -SRB™568/585, which belong to the most active cyclooctynes due to dibenzoannulation and introduction of an amide nitrogen in the strained ring system (k ∼ 0.36 M−1 s−1, Fig. 3A).30 The reaction was loaded onto a PAA-gel and analyzed by in-gel fluorescence (Fig. 3B).
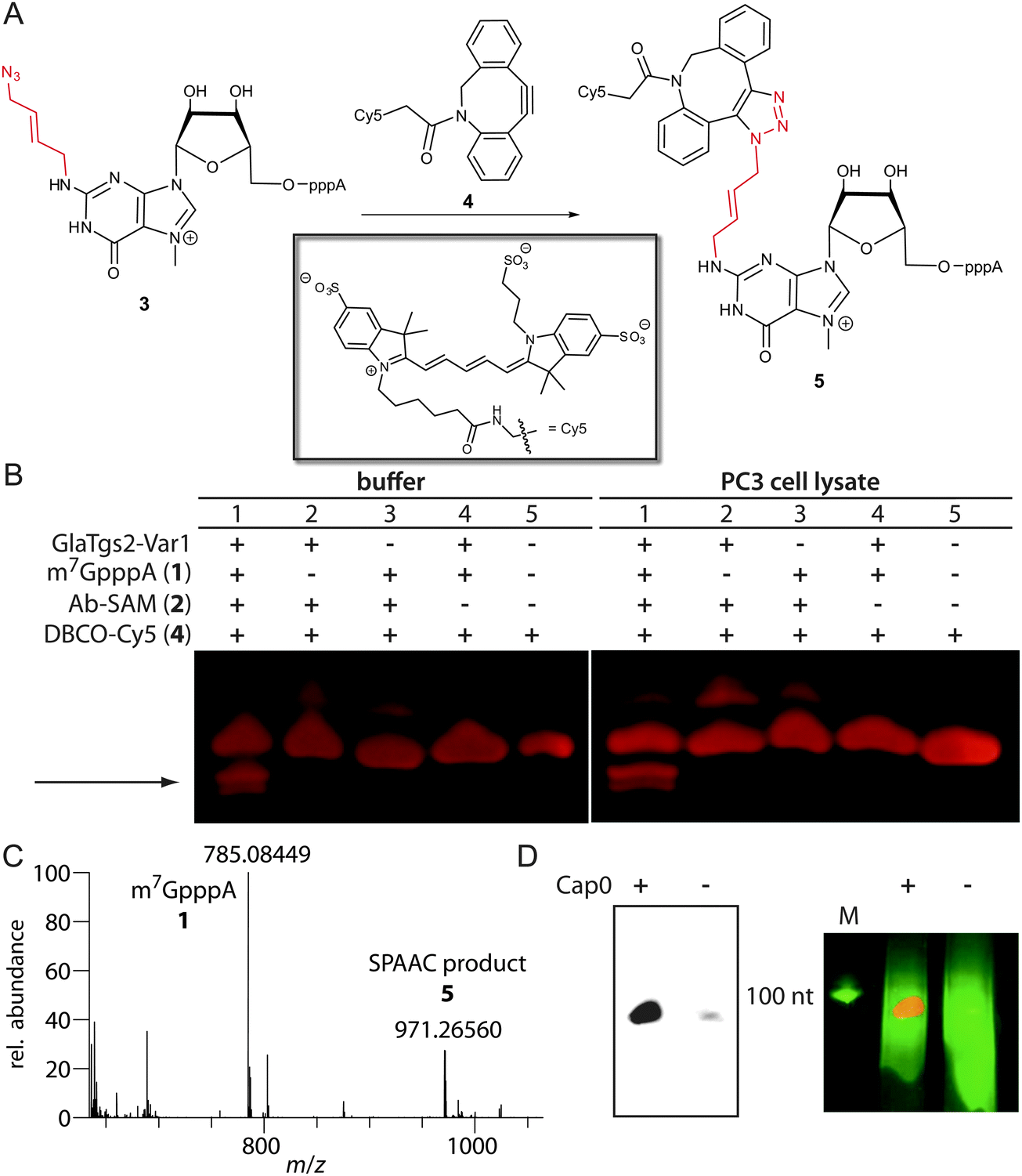 | ||
| Fig. 3 Labeling of enzymatically modified 5′-caps by SPAAC. (A) SPAAC reaction of N2-azidobutenyl-m7GpppA 3 with DBCO-Cy5™646/661 4 leading to modified minimal capped mRNA 5. (B) Analysis of chemo-enzymatic cap modification using in-gel fluorescence and PAGE. SPAAC was performed in PBS-buffer as well as in the PC3 cell lysate using DBCO-Cy5™646/661. Samples containing N2-4-azidobut-2-enyl-m7GpppA and DBCO-Cy5 reveal two new fluorescent bands (arrow). These bands were not present in controls, lacking either m7GpppA, enzyme or Ab-SAM. (C) ESI-Orbitrap-MS of the SPAAC reaction in the negative ion mode using DBCO-SRB™568/585 showing m7GpppA 1 (calculated for C21H28N10O17P3−: 785.08468 [M+ − 2H]−; found 785.08449) and the SPAAC product (calculated for C81H97N18O29P3S22−: 971.26616 [M+ − 3H]2−; found 971.26560). (D) Chemo-enzymatic labeling of in vitro transcribed and capped RNA (+) by SPAAC using DBCO-SRB™568/585. On the left SRB fluorescence was analyzed and on the right an overlay of SRB (red) and ethidiumbromide fluorescence (green) is shown. The negative control (−) contained non-capped RNA of the same length. | ||
Lane 5 shows DBCO-Cy5 alone that migrates into the gel, which is expected due to its negative charge. The appearance of a second band (lanes 1–3), which is absent in the control reaction without Ab-SAM (lane 4), may result from reaction of 2 with 4.
Lane 1 shows the reaction containing all components. Only in this reaction, two new fluorescent bands with a faster migration velocity appear (marked by arrow) that can be assigned to product 5. Formation of the SPAAC product using DBCO-SRB was confirmed by ESI-Orbitrap-MS (Fig. 3C). Appearance of two bands in the gel indicates that the triazole 1,4- and 1,5-regioisomers are formed.31 The faster migration suggests that the negative charge of the triphosphate in product 5 outperforms the additional molecular weight of 5 compared to 4. In comparison, the cap analog 1 alone migrates much faster than the obtained click product, which could be detected by UV-shadowing (Fig. S1 in the ESI†).
HPLC-based analyses using absorbance and fluorescence detection suggest that the SPAAC reaction leads to almost complete conversion of 3 to 5 (Fig. S3 in the ESI†).
To test whether our approach is also compatible with the complex environment of eukaryotic cells, we carried out the reaction in the lysate of PC3 cells, a human prostate cancer cell line. The reaction was successful and a similar band pattern compared to the reaction in PBS buffer was obtained (Fig. 3B). Again, the product bands for 5 were only formed if all components are present.
In a next step our chemo-enzymatic approach was used to label longer capped RNA molecules that are synonymous with eukaryotic mRNAs. A 106 nt long RNA was generated by in vitro transcription and capped using the vaccinia capping system. After the transfer reaction with 2 catalyzed by GlaTgs2-Var1 and the subsequent SPAAC reaction using DBCO-SRB™568/585, fluorescently labeled RNA could be detected by in-gel fluorescence (Fig. 3D). Non-capped RNA that was treated in the same way could only be visualized by ethidiumbromide staining.
In conclusion, we have developed a chemo-enzymatic approach for site-specific modification of 5′-capped RNAs that uses chemistry compatible with living cells. Our strategy is based on the enzymatically catalyzed modification of the mRNA cap by a distinct reporter group followed by a bioorthogonal click reaction. By harnessing the ability of the engineered GlaTgs2-Var1 to recognize an m7G-triphosphate at the 5′-end of the biopolymer RNA, the bioorthogonal azido group could be introduced and enables the attachment of functional moieties of choice. Using SPAAC we could fluorescently label the cap even in the complex environment of the eukaryotic cellular lysate. Moreover, we could demonstrate that our approach can be used to fluorescently label in vitro transcribed and capped RNAs. Since the cap is a hallmark of eukaryotic mRNAs, our chemo-enzymatic approach opens up a wide range of possible applications, such as isolation of mRNAs from total eukaryotic RNA or the selective labeling and subsequent visualization of mRNAs inside of living systems. To label specific mRNAs, GlaTgs2-Var1 could be covalently linked to proteins that bind RNAs sequence-specifically.
A.R. gratefully acknowledges financial support from the Emmy Noether-Programme of the DFG (RE 2796/2-1) and the Fonds der Chemischen Industrie. This work was partly supported by the Deutsche Forschungsgemeinschaft, DFG EXC 1003 Cells in Motion – Cluster of Excellence, Münster, Germany. J.M.H. thanks the Fonds der Chemischen Industrie for a doctoral fellowship. We acknowledge the MS facility of the Department of Chemistry, University of Hamburg (Dr Maria Trusch, Christine Christ, and Gaby Graack) for assistance with MALDI-TOF-MS and the mass spectrometry group of the Department of Chemistry and Pharmacy, University of Muenster (Dr Matthias Letzel), for assistance with ESI-Orbitrap-MS. We thank Prof. Hahn (University of Hamburg (Germany)) for providing the DNA template for transcription. We thank Prof. Zhaohui Zhou (Northeastern University, Boston, MA, USA) and Prof. Birgit Dräger (University of Halle (Germany)) for plasmids encoding LuxS and MTAN, respectively.
References
- M. Baker, Nat. Methods, 2012, 9, 787–790 CrossRef CAS.
- E. Bertrand, P. Chartrand, M. Schaefer, S. M. Shenoy, R. H. Singer and R. M. Long, Mol. Cell, 1998, 2, 437–445 CrossRef CAS PubMed.
- J. Koenig, S. Baumann, J. Koepke, T. Pohlmann, K. Zarnack and M. Feldbruegge, EMBO J., 2009, 28, 1855–1866 CrossRef CAS PubMed.
- D. Fusco, N. Accornero, B. Lavoie, S. M. Shenoy, J.-M. Blanchard, R. H. Singer and E. Bertrand, Curr. Biol., 2003, 13, 161–167 CrossRef CAS PubMed.
- I. Golding and E. C. Cox, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11310–11315 CrossRef CAS PubMed.
- C. Y. Jao and A. Salic, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15779–15784 CrossRef CAS PubMed.
- M. Grammel, H. Hang and N. K. Conrad, ChemBioChem, 2012, 13, 1112–1115 CrossRef CAS PubMed.
- H. Rao, A. A. Tanpure, A. A. Sawant and S. G. Srivatsan, Nat. Protoc., 2012, 7, 1097–1112 CrossRef CAS PubMed.
- Y. Motorin, J. Burhenne, R. Teimer, K. Koynov, S. Willnow, E. Weinhold and M. Helm, Nucleic Acids Res., 2011, 39, 1943–1952 CrossRef CAS PubMed.
- M. Tomkuviene, B. Clouet-d'Orval, I. Cerniauskas, E. Weinhold and S. Klimasauskas, Nucleic Acids Res., 2012, 40, 6765–6773 CrossRef CAS PubMed.
- E. Lecuyer, H. Yoshida, N. Parthasarathy, C. Alm, T. Babak, T. Cerovina, T. R. Hughes, P. Tomancak and H. M. Krause, Cell, 2007, 131, 174–187 CrossRef CAS PubMed.
- H. D. Lipshitz and C. A. Smibert, Curr. Opin. Genet. Dev., 2000, 10, 476–488 CrossRef CAS PubMed.
- K. C. Martin and A. Ephrussi, Cell, 2009, 136, 719–730 CrossRef CAS PubMed.
- G. J. Bassell and S. T. Warren, Neuron, 2008, 60, 201–214 CrossRef CAS PubMed.
- D. Schulz, J. M. Holstein and A. Rentmeister, Angew. Chem., Int. Ed., 2013, 52, 7874–7878 CrossRef CAS PubMed.
- D. C. Kennedy, C. S. McKay, M. C. B. Legault, D. C. Danielson, J. A. Blake, A. F. Pegoraro, A. Stolow, Z. Mester and J. P. Pezacki, J. Am. Chem. Soc., 2011, 133, 17993–18001 CrossRef CAS PubMed.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Org. Lett., 2008, 10, 3097–3099 CrossRef CAS PubMed.
- J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef CAS PubMed.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. Hendriks, F. P. Rutjes, J. C. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS PubMed.
- S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664–667 CrossRef CAS PubMed.
- S. T. Laughlin and C. R. Bertozzi, ACS Chem. Biol., 2009, 4, 1068–1072 CrossRef CAS PubMed.
- P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821–1826 CrossRef CAS PubMed.
- I. Singh, C. Freeman, A. Madder, J. S. Vyle and F. Heaney, Org. Biomol. Chem., 2012, 10, 6633–6639 CAS.
- K. N. Jayaprakash, C. G. Peng, D. Butler, J. P. Varghese, M. A. Maier, K. G. Rajeev and M. Manoharan, Org. Lett., 2010, 12, 5410–5413 CrossRef CAS PubMed.
- M.-L. Winz, A. Samanta, D. Benzinger and A. Jäschke, Nucleic Acids Res., 2012, 40, e78 CrossRef CAS PubMed.
- K. Islam, I. Bothwell, Y. Chen, C. Sengelaub, R. Wang, H. Deng and M. Luo, J. Am. Chem. Soc., 2012, 134, 5909–5915 CrossRef CAS PubMed.
- C. Dalhoff, G. Lukinavicius, S. Klimasauskas and E. Weinhold, Nat. Chem. Biol., 2006, 2, 31–32 CrossRef CAS PubMed.
- T. Suzuki, Y. Ikeuchi, A. Noma and Y. Sakaguchi, Methods Enzymol., 2007, 425, 211–229 CAS.
- M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. Rutjes, J. C. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC.
- F. Schoenebeck, D. H. Ess, G. O. Jones and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 8121–8133 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc01549e |
| This journal is © The Royal Society of Chemistry 2014 |