 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-innocent pyridyl nitrogens: unprecedented interconversion of N-bridgehead-thiadiazolium salts and thiatriazine in the generation of thiatriazinyl†
Alicea A.
Leitch
a,
Ilia
Korobkov
a,
Abdeljalil
Assoud
b and
Jaclyn L.
Brusso
*a
aDepartment of Chemistry and Centre for Catalysis Research and Innovation, University of Ottawa, 10 Marie Curie, Ottawa, ON K1N 6N5, Canada. E-mail: jbrusso@uottawa.ca
bDepartment of Chemistry, University of Waterloo, Waterloo, ON N2L 3G1, Canada
First published on 4th April 2014
Abstract
Condensation of N-2-pyridylimidoyl-2-pyridylamidine with S2Cl2 affords fused N-bridgehead-1,2,5-thiadiazolium salts, which can be converted to 3,5-bis(2-pyridyl)-4-hydro-1,2,4,6-thiatriazine (Py2TTAH). Oxidation of Py2TTAH with iodine yields the corresponding 1,2,4,6-thiatriazinyl radical, identified by EPR spectroscopy.
Over the past decade, much research has focused on the use of stable neutral radicals as building blocks for molecular conductors and magnetic materials.1,2 Their application as spin bearing ligands in coordination complexes has also been actively pursued.1,3 Within this context, the use of chelating heterocyclic neutral radicals is an attractive design strategy, as has been demonstrated by pyridyl functionalized verdazyls (1, Chart 1)4 and dithiadiazolyls (2).5 In principle, the 1,2,4,6-thiatriazinyl (TTA) framework,6 a neutral seven π-electron ring system, represents an ideal building block in the rational design of chelating spin bearing ligands.7 In particular, 3,5-bis(2-pyridyl)-1,2,4,6-thiatriazinyl (3; Py2TTA) would possess a chelating environment similar to that of 2,2′;6′,2′′-terpyridine (terpy); a tridentate ligand that has received a great deal of attention (e.g., close to 3000 publications in the last five years) due to its potential in a wide range of research areas (e.g., biomedical applications, catalysis, gas adsorption, magnetic materials, organic electronics, etc.).8 Given the immense interest in coordination complexes based on terpy, the generation of a structural mimic in which one of the pyridine rings is replaced by a TTA radical is appealing. Although phenyl functionalized TTA radicals are known, the reactivity of the pyridyl derivatives described here is profoundly different due to the presence of non-innocent pyridyl nitrogens, which can coordinate to sulphur and generate N-bridgehead-heterocycles.9 In that regard, the unprecedented but necessary interconversion of an N-bridgehead-1,2,5-thiadiazolium salt to a 1,2,4,6-thiatriazine (TTAH) precedes the generation of 3. Herein, the synthetic sequence and molecular structures of the intermediates will be presented along with EPR characterization of the 3,5-bis(2-pyridyl)-1,2,4,6-thiatriazinyl radical (3).
 | ||
| Chart 1 | ||
The first report of a TTA radical was described by Markovskii et al. using EPR spectroscopy.10 Since then, both symmetrically and asymmetrically substituted TTA radicals have been prepared, most of which are, at least partly, functionalized with aryl groups.6,11–13 Known preparative routes include the reaction of amidines with S3N3Cl36,11 or condensation of imidoylamidine hydrochlorides with excess SCl2,13,14 followed by reduction with Ph3Sb. Our synthetic sequence followed a similar route, as outline in Scheme 1, in which N-2-pyridylimidoyl-2-pyridylamidine (4), prepared from reaction of 2-cyanopyridine with NH3(g), was treated with S2Cl2. This reaction did not, however, generate the anticipated 3,5-bis(2-pyridyl)-1-chloro-1,2,4,6-thiatriazine. Instead, the condensation afforded [5][Cl]·HCl, a dication containing N-bridgehead-1,2,5-thiadiazolium and pyridinium moieties. This material was isolated as an insoluble chloride salt, which was metathesized using trimethylsilyl triflate to a soluble triflate salt ([5][OTf]·HOTf). Crystallization from acetonitrile (MeCN) afforded colourless needles suitable for X-ray analysis, the results of which are shown in Fig. 1a.‡ The planarity of [5][OTf]·HOTf (mean deviation of 0.0746 Å from the 18 atom framework), coupled with its short C–N bond lengths, suggests some degree of resonance delocalization along the central N–C–N–C–N backbone.
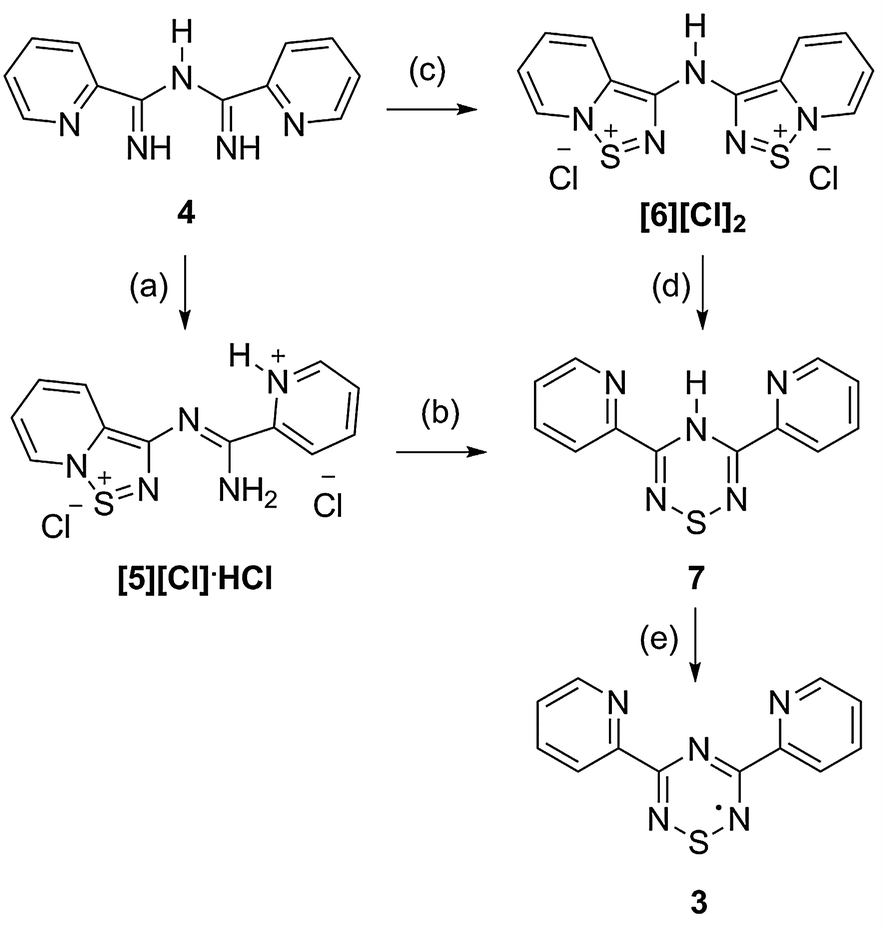 | ||
| Scheme 1 Synthesis of bis(2-pyridyl)-1,2,4,6-thiatriazinyl radical. Reagents and conditions: (a) (i) S2Cl2, MeCN, RT; (ii) 100 °C, 10−2 mmHg; (b) 140 °C, 10−2 mmHg; (c) S2Cl2, MeCN, reflux; (d) Ph3Sb, MeCN, reflux; (e) DMAP, I2, DCM. | ||
 | ||
| Fig. 1 ORTEP drawings (50% thermal ellipsoids) of (a) [5][OTf]·HOTf and (b) [6][OTf]2. Anions have been removed for clarity. | ||
Initially, isolation of [5][Cl]·HCl was surprising as we were expecting to generate 3,5-bis(2-pyridyl)-1-chloro-1,2,4,6-thiatriazine. Clearly the reactivity of the pyridyl derivatives described here is in marked contrast to the previously reported phenyl functionalized TTA analogues. This is attributed to the ability of the pyridyl nitrogen atoms to coordinate to sulphur. To our knowledge, the only other example of such an interaction was reported by Rawson et al.9 Given the availability of two pyridyl substituents, the possibility of generating a bis(N-bridgehead-1,2,5-thiadiazolium) dication [6]2+ was considered. To that end, N-2-pyridylimidoyl-2-pyridylamidine (4) was treated with excess S2Cl2 at reflux, affording [6]2+ as an insoluble chloride salt that gave a distinctly different IR spectrum compared to [5][Cl]·HCl. To confirm the identity of [6]2+, it was converted into the corresponding triflate salt, [6][OTf]2, by treatment with trimethylsilyl triflate. Colourless needles suitable for structural analysis were obtained by crystallization from MeCN (Fig. 1b), demonstrating that [6]2+ is comprised of two nearly identical N-bridgehead-1,2,5-thiadiazolium moieties linked together by a central nitrogen atom, which are twisted with respect to one another by an angle of 33.26(4)°.
With [6]2+ in hand, a two-electron reduction could afford a diradical or lead to ring opening, as proposed by Rawson.9 In our hands, treatment of [6][Cl]2 with Ph3Sb at reflux generated a deep burgundy solution which, upon cooling, afforded deep red needles of 3,5-bis(2-pyridyl)-4-hydro-1,2,4,6-thiatriazine (7); the structural identity of which was confirmed by X-ray analysis (Fig. 2). This closed-shell molecule is bent along the N2–S1 axis with an angle of 153.83(3)° between the two halves of the framework. This, coupled with the short C1–N1 and C2–N3 bond lengths, indicates an antiaromatic structure, as is expected for TTAH.15
 | ||
| Fig. 2 ORTEP drawings (50% thermal ellipsoids) of 7 viewed from (a) above and (b) side of the molecular framework. | ||
Alternatively, 7 can also be prepared via thermolysis of [5][Cl]·HCl at 140 °C in vacuo or at reflux in chlorobenzene. It is therefore apparent that the key intermediates in the formation of pyridine functionalized TTA heterocycles are the N-bridgehead-1,2,5-thiadiazolium cations. Furthermore, treatment of 7 with a proton source (e.g., HCl(g)) regenerates [5][Cl]·HCl. Thus, thermal treatment of [5][Cl]·HCl causes rearrangement to the thiatriazine, whereas the presence of acid favours TTAH ring opening and generation of [5][Cl]·HCl. This unprecedented interconversion of the N-bridgehead-1,2,5-thiadiazolium and TTAH may be monitored visually, as [5][Cl]·HCl is a colourless solid and 7 is deep red. Accordingly, this system may have potential in thermo/acidochromic applications.
Regardless of how pyridine functionalized TTAH is prepared, its conversion to the corresponding radical, 3,5-bis(2-pyridyl)-1,2,4,6-thiatriazinyl (3), can be effected by oxidation. To that end, treatment of 7 with half an equivalent of iodine in the presence of base (e.g., 4-dimethylaminopyridine) yields a dark red solution that exhibits a strong and persistent EPR signal (Fig. 3) whose appearance is consistent with TTA radicals bearing electron-withdrawing substituents that polarize spin density away from the N–S–N region of the TTA core.12,16 Indeed the EPR spectrum of 3 is virtually identical to that reported for 3,5-bis(p-nitrophenyl)-1,2,4,6-thiatriazinyl (cf. g = 2.0055; aN = 0.372 mT; aN = 0.427 mT).12 Accordingly, the observed signal consists of a complex multiplet that can be simulated using a model based on hyperfine coupling to two equivalent and one unique 14N nuclei (experimentally derived constants: aN = 0.377 mT; aN = 0.438 mT; calculated coupling constants: aN = 0.347 mT; aN = 0.420 mT).
 | ||
| Fig. 3 (a) Experimental and simulated EPR spectrum of 3 in DCM (g = 2.0068; SW = 3.5 mT; LW = 0.024 mT). (b) Experimentally derived and UB3LYP/EPR-II/6-31G(d)//UB3LYP/6-311G(d,p) calculated (in parenthesis) coupling constants aN (in mT). (c) UB3LYP/6-311G(d,p) singly occupied molecular orbital. | ||
Based on this study, it is clear the presence of pyridyl substituents in the development of sulphur/nitrogen heterocycles has a significant impact on reaction pathways. In particular, the coordinating ability of the pyridine nitrogen atoms, and the apparent proclivity of pyridyl ligands to form N-bridgehead-heterocycles, is an important finding and holds potential in the design of novel open and closed shell heterocyclic compounds. The synthetic challenges associated with non-innocent pyridyl nitrogens can be overcome, as demonstrated here in the preparation of the 3,5-bis(2-pyridyl)-1,2,4,6-thiatriazinyl radical (3). In conclusion, not only do we anticipate rich coordination chemistry for this radical acting as a multidentate chelating ligand, we also foresee the possibility of developing new radical ion and biradical systems from controlled reduction of [6]2+ and related compounds.
The authors thank the University of Ottawa, the Canada Foundation for Innovation and National Sciences and Engineering Research Council of Canada for financial support and are grateful to Dr Aaron Mailman for recording the EPR spectrum of 3.
Notes and references
- (a) R. G. Hicks, Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds, John Wiley & Sons, Ltd., Wiltshire, 2010 Search PubMed; (b) I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303 RSC.
- (a) R. G. Hicks, Nat. Chem., 2011, 3, 189 CrossRef CAS PubMed; (b) A. W. Cordes, R. C. Haddon and R. T. Oakley, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 673 CrossRef CAS; (c) A. Jankowiak, D. Pociecha, J. Szczytko, H. Monobe and P. Kaszynski, J. Am. Chem. Soc., 2012, 134, 2465 CrossRef CAS PubMed; (d) J. M. Rawson, A. Alberola and A. Whalley, J. Mater. Chem., 2006, 16, 2560 RSC; (e) J. W. L. Wong, A. Mailman, K. Lekin, S. M. Winter, W. Yong, J. Zhao, S. V. Garimella, J. S. Tse, R. A. Secco, S. Desgreniers, Y. Ohishi, F. Borondics and R. T. Oakley, J. Am. Chem. Soc., 2014, 136, 1070 CrossRef CAS PubMed.
- (a) K. E. Preuss, Dalton Trans., 2007, 2357 RSC; (b) M. Sorai, Y. Nakazawa, M. Nakano and Y. Miyazaki, Chem. Rev., 2013, 113, PR41 CrossRef PubMed; (c) C. Y. Ang, R. T. Boere, L. Y. Goh, L. L. Koh, S. L. Kuan, G. K. Tan and X. Yu, Chem. Commun., 2006, 4735 RSC; (d) K. Awaga, K. Nomura, H. Kishida, W. Fujita, H. Yoshikawa, M. M. Matsushita, L. Hu, Y. Shuku and R. Suizu, Bull. Chem. Soc. Jpn., 2014, 87, 234 CrossRef CAS.
- (a) C. W. Johnston, S. D. J. McKinnon, B. O. Patrick and R. G. Hicks, Dalton Trans., 2013, 42, 16829 RSC; (b) S. D. J. McKinnon, B. O. Patrick, A. B. P. Lever and R. G. Hicks, Chem. Commun., 2010, 46, 773 RSC.
- (a) N. G. R. Hearns, R. Clerac, M. Jennings and K. E. Preuss, Dalton Trans., 2009, 3193 RSC; (b) J. Britten, N. G. R. Hearns, K. E. Preuss, J. F. Richardson and S. Bin-Salamon, Inorg. Chem., 2007, 46, 3934 CrossRef CAS PubMed; (c) N. G. R. Hearns, E. M. Fatila, R. Clerac, M. Jennings and K. E. Preuss, Inorg. Chem., 2008, 47, 10330 CrossRef CAS PubMed; (d) N. G. R. Hearns, K. E. Preuss, J. F. Richardson and S. Bin-Salamon, J. Am. Chem. Soc., 2004, 126, 9942 CrossRef CAS PubMed.
- A. W. Cordes, P. J. Hayes, P. D. Josephy, H. Koenig, R. T. Oakley and W. T. Pennington, J. Chem. Soc., Chem. Commun., 1984, 1021 RSC.
- J. Wu, PhD thesis, University of Guelph, 2008.
- (a) E. C. Constable, Chem. Soc. Rev., 2007, 36, 246 RSC; (b) E. A. Medlycott and G. S. Hanan, Chem. Soc. Rev., 2005, 34, 133 RSC; (c) P. R. Andres and U. S. Schubert, Adv. Mater., 2004, 16, 1043 CrossRef CAS PubMed; (d) R. Shunmugam, G. J. Gabriel, K. A. Aamer and G. N. Tew, Macromol. Rapid Commun., 2010, 31, 784 CrossRef CAS PubMed.
- C. E. Bacon, D. J. Eisler, R. L. Melen and J. M. Rawson, Chem. Commun., 2008, 4924 RSC.
- L. N. Markovskii, P. P. Kornuta, L. S. Kachkovskaya and O. M. Polumbrik, Sulfur Lett., 1983, 1, 3 Search PubMed.
- P. J. Hayes, R. T. Oakley, A. W. Cordes and W. T. Pennington, J. Am. Chem. Soc., 1985, 107, 1346 CrossRef CAS.
- R. T. Boere, R. T. Oakley, R. W. Reed and N. P. C. Westwood, J. Am. Chem. Soc., 1989, 111, 1180 CrossRef CAS.
- R. T. Boere, T. L. Roemmele and X. Yu, Inorg. Chem., 2011, 50, 5123 CrossRef CAS PubMed.
- R. T. Oakley, R. W. Reed, A. W. Cordes, S. L. Craig and J. B. Graham, J. Am. Chem. Soc., 1987, 109, 7745 CrossRef CAS.
- R. T. Boere, A. W. Cordes, P. J. Hayes, R. T. Oakley, R. W. Reed and W. T. Pennington, Inorg. Chem., 1986, 25, 2445 CrossRef CAS.
- R. T. Boere and T. L. Roemmele, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 875 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, crystallographic details and computational studies. CCDC 986942–986944. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc01433b |
‡ Crystal data at 200(2) K for [5][OTf]·HOTf: C14H11F6N5O6S3, M = 555.46, triclinic, a = 9.1987(3) Å, b = 9.8364(3) Å, c = 11.7652(4) Å, α = 94.5844(16)°, β = 102.6573(17)°, γ = 92.0173(17)°, V = 1033.82(6) Å3, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, 5084 reflections measured, 3959 unique (Rint = 0.0288). The final wR(F2) was 0.1220 (all data). Crystal data at 296(2) K for [6][OTf]2: C14H9F6N5O6S4, M = 585.50, triclinic, a = 8.4770(3) Å, b = 10.1380(3) Å, c = 12.6490(4) Å, α = 88.7192(17)°, β = 78.5886(16)°, γ = 83.6975(17)°, V = 1059.12(6) Å3, space group P, Z = 2, 4339 reflections measured, 3639 unique (Rint = 0.0188). The final wR(F2) was 0.1195 (all data). Crystal data at 200(2) K for 7: C12H9N5S, M = 255.30, monoclinic, a = 13.6633(4) Å, b = 3.84480(10) Å, c = 22.7022(7) Å, β = 106.326(2)°, V = 1144.52(6) Å3, space group P21/c, Z = 4, 2799 reflections measured, 2174 unique (Rint = 0.0377). The final wR(F2) was 0.1183 (all data). , Z = 2, 5084 reflections measured, 3959 unique (Rint = 0.0288). The final wR(F2) was 0.1220 (all data). Crystal data at 296(2) K for [6][OTf]2: C14H9F6N5O6S4, M = 585.50, triclinic, a = 8.4770(3) Å, b = 10.1380(3) Å, c = 12.6490(4) Å, α = 88.7192(17)°, β = 78.5886(16)°, γ = 83.6975(17)°, V = 1059.12(6) Å3, space group P, Z = 2, 4339 reflections measured, 3639 unique (Rint = 0.0188). The final wR(F2) was 0.1195 (all data). Crystal data at 200(2) K for 7: C12H9N5S, M = 255.30, monoclinic, a = 13.6633(4) Å, b = 3.84480(10) Å, c = 22.7022(7) Å, β = 106.326(2)°, V = 1144.52(6) Å3, space group P21/c, Z = 4, 2799 reflections measured, 2174 unique (Rint = 0.0377). The final wR(F2) was 0.1183 (all data). |
| This journal is © The Royal Society of Chemistry 2014 |