 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the hydrates of codeine phosphate: the remarkable influence of hydrogen bonding on the crystal size†
Tomče
Runčevski
*a,
Gjorgji
Petruševski
b,
Petre
Makreski
c,
Sonja
Ugarkovic
b and
Robert E.
Dinnebier
a
aMax Planck Institute for Solid State Research, Heisenbergstrasse 1, 70569 Stuttgart, Germany. E-mail: t.runcevski@fkf.mpg.de
bResearch & Development, ALKALOID AD, Aleksandar Makedonski 12, 1000 Skopje, Macedonia
cInstitute of Chemistry, Faculty of Natural Sciences and Mathematics, SS. Cyril and Methodius University, Arhimedova 5, 1000 Skopje, Macedonia
First published on 6th March 2014
Abstract
Codeine phosphate forms three hydrates and two anhydrates. The sesquihydrate and hemihydrate, which differ by one water molecule, are stable at room temperature. The influence of this molecule on the internal crystal structure and how it translates into the external crystal shape are reported.
Codeine is a natural alkaloid, which is extracted from the opium poppy plant. It is a well-known narcotic, analgesic, antitussive and anti-diarrhoeal active pharmaceutical ingredient (API) and has a long history, for example, in the treatment of mild to moderate pain, coughs, diarrhoea, and irritable bowel syndrome.1 It is such a widely used narcotic that it can be found in the WHO Model List of Essential Drugs. Codeine is frequently marketed in the form of its phosphate salt (COP, Chart 1), only two hydrates of which are commercially used in the preparation of pharmaceutical formulations: the sesquihydrate (COP-S; 1.5 water equivalents per codeine cation) and the hemihydrate (COP-H; 0.5 water equivalents). Recently, thermoanalytical studies (TGA/DSC) were published, revealing that, upon heating, COP-S transforms into an unstable monohydrate (COP-M), which subsequently converts into COP-H. Heating above 100 °C leads to the formation of two anhydrous forms (COP-AI and COP-AII).2
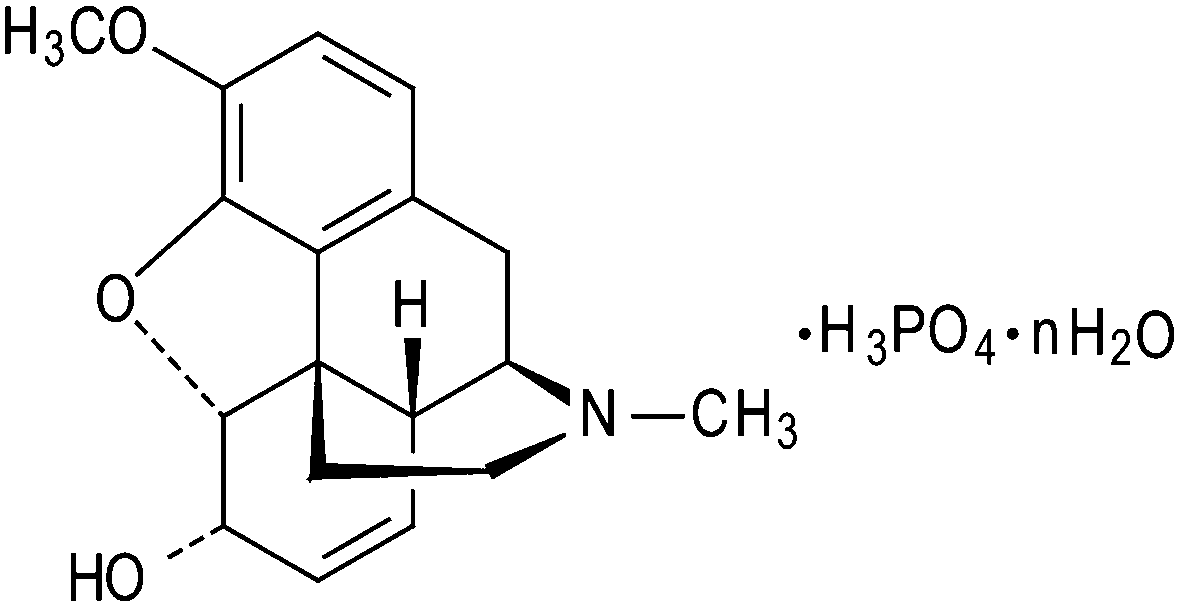 | ||
| Chart 1 Structural formula of the codeine phosphate n-hydrate. | ||
Even though COP is an important API, there are many unanswered questions regarding its solid state properties. Surprisingly, especially given the fact that it is an API that has been used in the pharmaceutical industry for more than half a century, the published crystallographic research is scarce. The only crystal structure detailed in the literature is that of COP-H.3 Among the most significant reasons for such poor crystallographic knowledge of COP is its crystallization behaviour; it yields single crystals of COP-H, but a polycrystalline bulk of COP-S (Fig. 1). It is known that changes in the crystal structure alter the physical properties of a solid, including the crystallization behaviour.4 The effect of composition on the crystallization behaviour displayed here is rather impressive as is governed by the one water molecule difference between COP-S and COP-H. The ability of the water molecule to change the crystal structure (e.g. via hydrogen bonding networks) is recognised,4 and the COP hydrates stand out as extreme examples of how it translates into the physical size and shape of the crystal. Moreover, when dealing with APIs, the physics of the solid is very important, as it might affect a number of properties, including (but not limited to) toxicity, bioavailability, chemical stability or shelf life.5 Therefore, an understanding of the role of one additional water molecule in how the COP crystal grows is highly desirable.
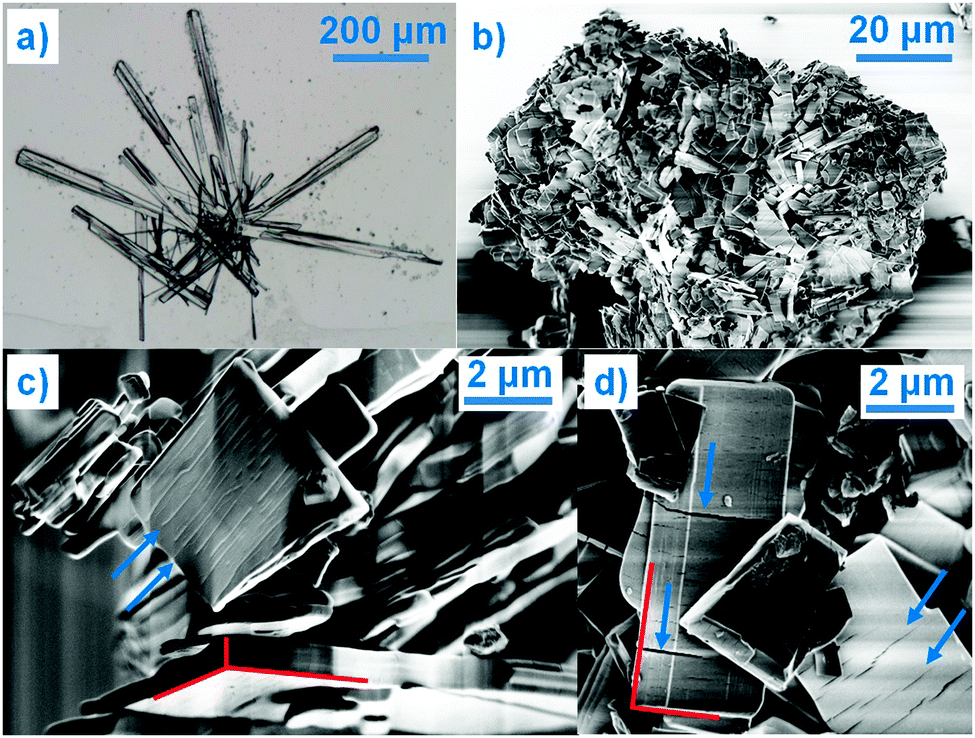 | ||
| Fig. 1 (a) Optical microscopy image of COP-H single crystals, and (b–d) SEM images of COP-S polycrystalline particles (the red lines are a guide to the eye for the microcrystals habit whereas the blue arrows indicate the cracks perpendicular to the largest crystal axis). | ||
In the course of the present research, as well as during previous studies on the solid state transformations and solvatomorphism,2 recrystallization of COP in different solvents always leads to single crystals of COP-H and powder samples of COP-S. Given the polycrystalline nature of COP-S, here, its crystal structure was solved using X-ray powder diffraction (XRPD), which is an emerging technique for structure solutions of pharmaceuticals.6 Indexing was performed using first principles by the iterative use of singular value decomposition, followed by Pawley fitting and the simulated annealing approach to solve the crystal structure, which was later refined by the Rietveld method, confirming the solution and, in addition, providing information on the microstructure.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ‡ The asymmetric unit of COP-S is composed of two codeine cations adopting the characteristic T conformation known from related compounds of the opiate family.3 One slight difference in the crystal structures of the codeine cations in COP-S and COP-H is the orientation of the methoxy group. The crystal packing of the cations is similar to the one in COP-H, as shown in Fig. 2(a–d). The similar packing of the codeine cations cannot solely account for the difference in crystallization behaviour. Irrespective of being similarly packed, independent codeine units are linked differently to other constituents of the asymmetric unit. In COP-H, two cations are linked together through (codeine)–O–H⋯O–(codeine) hydrogen bonds, employing the hydroxy and methoxy groups, the hydroxy group is additionally bonded to the water molecule and moreover, both codeine cations are bonded to the phosphate anions via (codeine)–N–H⋯O–(phosphate) hydrogen bonds (Fig. 2(e)).3 The hydrogen bonding scheme of COP-S differs significantly from that of COP-H. In one of the codeine cations, the protonated amino group and the hydroxy group are both bonded to water molecules, whereas in the other cation the same groups are bonded to the oxygen atoms of the phosphate anions (Fig. 2(f)).
‡ The asymmetric unit of COP-S is composed of two codeine cations adopting the characteristic T conformation known from related compounds of the opiate family.3 One slight difference in the crystal structures of the codeine cations in COP-S and COP-H is the orientation of the methoxy group. The crystal packing of the cations is similar to the one in COP-H, as shown in Fig. 2(a–d). The similar packing of the codeine cations cannot solely account for the difference in crystallization behaviour. Irrespective of being similarly packed, independent codeine units are linked differently to other constituents of the asymmetric unit. In COP-H, two cations are linked together through (codeine)–O–H⋯O–(codeine) hydrogen bonds, employing the hydroxy and methoxy groups, the hydroxy group is additionally bonded to the water molecule and moreover, both codeine cations are bonded to the phosphate anions via (codeine)–N–H⋯O–(phosphate) hydrogen bonds (Fig. 2(e)).3 The hydrogen bonding scheme of COP-S differs significantly from that of COP-H. In one of the codeine cations, the protonated amino group and the hydroxy group are both bonded to water molecules, whereas in the other cation the same groups are bonded to the oxygen atoms of the phosphate anions (Fig. 2(f)).
 | ||
| Fig. 2 Crystal packing of COP-H shown in (a) and (c) and COP-S in (b) and (d). Hydrogen bonding of the codeine cations of (e) COP-H and (f) COP-S. The extended hydrogen bonding network of phosphate anions and water molecules of (g) COP-H and (h) COP-S. The unit cell content of COP-H is doubled for better comparison with COP-S. | ||
The crucial difference between the COP-H and COP-S structures is the hydrogen-bonding network of the phosphate anions and water molecules. The adjacent phosphate anions of COP-H are pairwise joined together by hydrogen bonds to build an extended ribbon chain on which the water molecules and codeine cations are attached (Fig. 2(g)). The presence of an additional water molecule leads to the completely different hydrogen bonding scheme in COP-S, where a complex 2D layer is formed (Fig. 2(h)).§ The phosphate anions are joined pairwise, additionally linking to water molecules as well as the hydroxy and protonated amino groups of the codeine cations. Three crystallographically different water molecules are present, two of them bonded to the codeine cations and one only participating in the formation of the phosphate–water 2D layer. The differences in the hydrogen bonding motifs are clearly reflected in the IR spectra.† As previously observed,2 the spectrum of COP-H exhibits two sharp peaks (3503 and 3460 cm−1) and two shoulders (3540 and 3401 cm−1) ascribed to mixed N–H and O–H vibrations. The extensive hydrogen bonding observed in the structure of COP-S helped to explain the large redshift, manifested by the appearance of a broad band with superimposed shoulders (∼3270, 3457 and 3500 cm−1).
The disparity in the crystal structures of COP-H and COP-S can be related to the differences in their crystallization behaviour and crystal shape. As previously indicated,3 a systematic comparison of the phosphate ribbon chain motif in COP-H with related structures points out that this very simple synthon is favoured by the packing of dihydrogen phosphate anions. Thus, the propagation of the chain and attached codeine cations from the surrounding solution forms druses of single crystals with the prismatic habit. With careful evaporation of the solvent, crystals of COP-H larger than 500 μm in length can be easily grown (Fig. 1(a)). In contrast, COP-S crystallizes as extensively cracked prismatic crystals rarely exceeding a few micrometres in size (Fig. 1(b–d)). Rietveld refinements indicated that average domain sizes range within 0.1 and 0.15 μm.‡ It is known that the crystal growth is heavily affected by the presence of defects in the structure, which often cause growth termination. The introduction of such defects in the simple phosphate chains in COP-H is less likely as compared to the complicated 2D network in COP-S, where the absence and/or displacement of, for example, a single H2O molecule, can severely disrupt the 2D structure. Interestingly, this 2D layer is packed perpendicularly to the largest unit cell axis (Fig. 2(b and d)), which possibly correlates to the cracks in the microcrystals, perpendicular to the longest crystal axis (Fig. 1(c and d)).
The well-defined, 2D hydrogen-bonded phosphate–water network, on which the codeine cations are attached, ensures the very stable crystal packing of COP-S, which is stable at room temperature (RT) and under both ambient and saturated humidity conditions.2aCOP-S was proved to be the highest hydrate, with 1.5 water molecules per codeine cation. The study of its thermally-induced process was revisited, complementing the published thermoanalytical data (summarized in Fig. 3(a))2 with in situ crystallographic analysis. The aim of this was to detect the unstable COP-M form and to firmly establish the dehydration scheme of this pharmaceutically important compound. The DSC curve (Fig. 3(a)) indicated that four endothermic transitions before thermal decomposition occurred (starting at ∼240 °C, as shown by the TG and DTG curves). The second peak visible on the DSC curve (at 99.9 °C) was not detected in the DTG or c-DTA analyses. To confirm that this low-energy effect is not due to the experimental error or an artefact, temperature-resolved XRPD data were collected in situ (Fig. 3(b)).¶ The changes in the scattered X-ray intensity of COP-S as a function of the diffraction angle and temperature nicely complement the DSC results, showing four transitions below 210 °C. The first three thermal effects are assigned to the dehydration sequence: COP-S → COP-M → COP-H → COP-AI, followed by one polymorphic transition, COP-AI → COP-AII, before total degradation takes place (Fig. 3c). An interesting observation is that, upon heating, unlike all other forms, COP-AII exhibits a large and continuous diffraction peak shift towards higher angles, indicating significant unit cell positive thermal expansion.
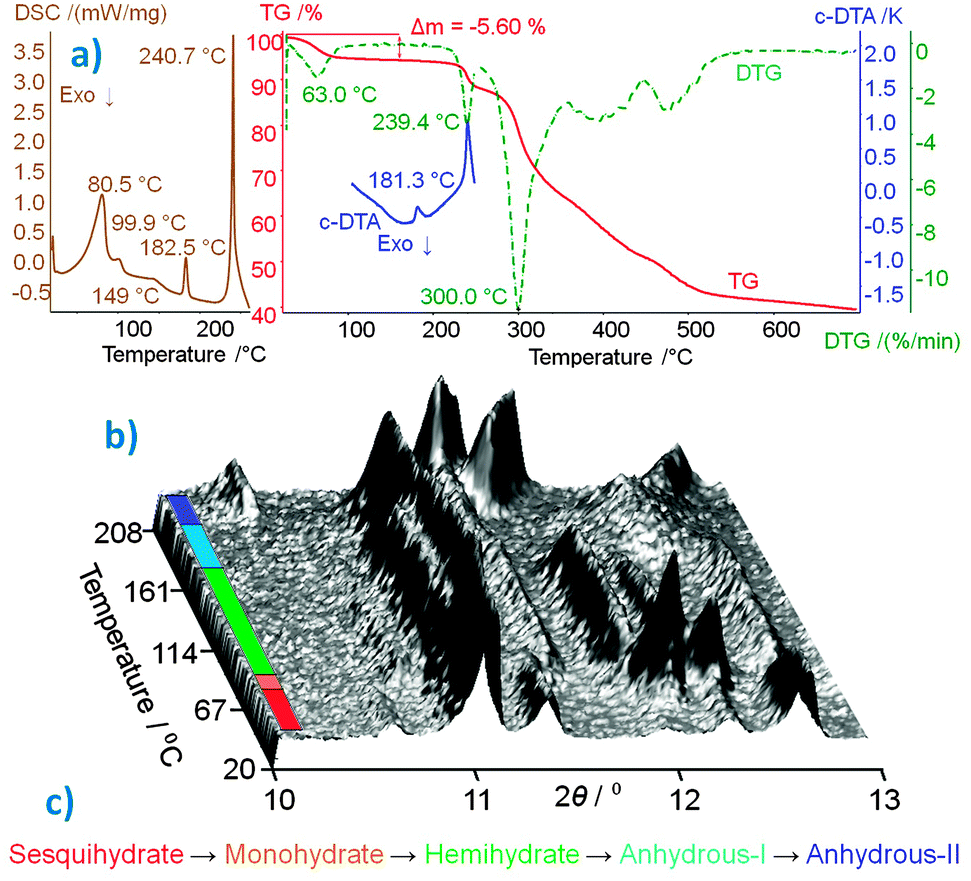 | ||
| Fig. 3 (a) Thermoanalytical characterization curves of COP-S.2 (b) 3D projection of scattered X-ray intensity of COP-S presented as a function of the diffraction angle and temperature, the different phases are colour coded according to the (c) dehydration/polymorphism sequence. | ||
In summary, COP crystallizes as two hydrates at RT, sesquihydrate COP-S (1.5 H2O) and hemihydrate COP-H (0.5 H2O), whereas the intermediate COP-M form (1 H2O) is unstable. At high temperatures, two anhydrous polymorphs are produced (COP-AI and COP-AII). The presence of 1.5 H2O molecules per codeine cation leads to a stable form at RT and at ambient and saturated humidities. Losing 0.5 H2O molecules per codeine cation equivalent results in instability (COP-M) and a further loss of 0.5 H2O molecules once again stabilizes the structure (COP-H). COP-S and COP-H have similarly packed codeine cations and differ only by one water molecule per cation. However, this single water molecule makes a huge difference in the hydrogen bonding network. This detail of the internal crystal structure highly influences the external crystal shape and stability. COP-H forms single crystals, whereas COP-S crystallizes as cracked microcrystals with domains as small as 0.1 μm.
The efforts of Mr Wied, Mrs Kuševska, Mrs Popovic and Dr Nuss in making the international transport of the drug possible are very much appreciated. Mrs Knöller is acknowledged for recording the SEM images and Mr Beardsworth for proof-reading the manuscript. TR thanks the IMPRS-CMS. The authors thank the anonymous reviewers for the valuable comments and suggestions which significantly improve the quality of this communication.
Notes and references
- (a) A. J. Atkinson, H. F. Adler and A. C. Ivy, J. Am. Med. Assoc., 1943, 121, 646 CrossRef; (b) N. B. Eddy, H. Friebel, K.-J. Hahn and H. Halbach, Bull. W. H. O., 1968, 38, 673 CAS; (c) N. B. Eddy, H. Friebel, K.-J. Hahn and H. Halbach, Bull. W. H. O., 1969, 40, 425 CAS.
- (a) G. Petruševski, S. Ugarkovic and P. Makreski, J. Mol. Struct., 2011, 993, 328 CrossRef PubMed; (b) G. Petruševski, M. Kajdžanoska, S. Ugarkovic, I. Micovski, G. Bogoeva-Gaceva, G. Jovanovski and P. Makreski, Vib. Spectrosc., 2012, 63, 60 CrossRef PubMed.
- C. Langes, T. Gelbrich, U. J. Griesser and V. Kahlenberg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, 419 Search PubMed.
- (a) J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, Oxford, 2002 Search PubMed; (b) G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS; (c) M. Wenger and J. Bernstein, Cryst. Growth Des., 2008, 8, 1595 CrossRef CAS; (d) C. Näther, I. Jess, P. G. Jones, C. Taouss and N. Teschmit, Cryst. Growth Des., 2013, 13, 1676 CrossRef; (e) D. Braga, F. Grepioni and L. Maini, Chem. Commun., 2010, 46, 6232 RSC; (f) C. Janiak, Dalton Trans., 2003, 2781 RSC; (g) C. B. Aakeroy, M. E. Fasulo and J. Desper, Mol. Pharmaceutics, 2007, 4, 317 CrossRef PubMed; (h) B. Rodríguez-Spong, C. P. Price, A. Jayasankar, A. J. Matzger and N. Rodríguez-Hornedo, Adv. Drug Delivery Rev., 2004, 56, 241 CrossRef PubMed; (i) M. R. Caira, Mol. Pharmaceutics, 2007, 4, 310 CrossRef CAS PubMed; (j) A. Othman, J. S. O. Evans, I. Radosavljevic Evans, R. K. Harris and P. Hodgkinson, J. Pharm. Sci., 2007, 69, 1380 CrossRef PubMed.
- (a) S. R. Byrn, R. R. Pfeiffer and J. G. Stowell, Solid State Chemistry of Drugs, SSCI Inc., West Lafayette, IN, 2nd edn, 1999 Search PubMed; (b) S. L. Morissette, O. Almarsson, M. L. Peterson, J. F. Remenar, M. J. Read, A. V. Lemmo, S. Ellis, M. J. Cima and C. R. Gardner, Adv. Drug Delivery Rev., 2004, 56, 275 CrossRef CAS PubMed; (c) N. Shan and M. J. Zaworotko, Drug Discovery Today, 2008, 13, 440 CrossRef CAS PubMed; (d) J. Chen, B. Sarma, J. M. B. Evans and A. S. Myerson, Cryst. Growth Des., 2011, 11, 887 CrossRef CAS; (e) O. Almarsson and M. J. Zaworotko, Chem. Commun., 2004, 1889 RSC.
- (a) R. E. Dinnebier, P. Sieger, H. Nar, K. Shankland and W. I. F. David, J. Pharm. Sci., 2000, 89, 1465 CrossRef CAS; (b) K. Sugimoto, R. E. Dinnebier and M. Zakrezewski, J. Pharm. Sci., 2007, 96, 3316 CrossRef CAS PubMed; (c) L. Vella-Zarb, R. E. Dinnebier and U. Baisch, Cryst. Growth Des., 2013, 13, 4402 CrossRef CAS; (d) K. D. M. Harris and E. Y. Cheung, Chem. Soc. Rev., 2004, 33, 526 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Description of sample preparation, Crystallographic details and the Rietveld plot of COP-S. CCDC 988910, FTIR spectra. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc01430h |
| ‡ Crystallographic details of COP-S: 2θ range 3–66°, X-ray radiation with λ = 1.540596 Å, data collection time of 24 h. Space group P212121, unit cell: a = 33.4761(7) Å, b = 16.0612(3) Å, c = 7.1921(2) Å; RBragg = 0.0225, Rexp = 0.0099, Rwp = 0.0348, Rp = 0.0285, 89 refined parameters. |
| § For measuring the hydrogen bonding, the intermolecular O–O distances were studied and the values are deposited in the CIF file. |
| ¶ In situ XRPD patterns were collected at each degree upon heating, in a 2θ range of 10–13°, λ = 1.540596 Å, with a data collection time of 2 min. |
| This journal is © The Royal Society of Chemistry 2014 |