 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA triple-channel lab-on-a-molecule for triple-anion quantification using an iridium(III)–imidazolium conjugate†
Kun
Chen
and
Michael
Schmittel
*
Center for Micro- and Nanochemistry and Engineering, Organische Chemie, Universität Siegen, Adolf-Reichwein-Strasse, D-57068, Siegen, Germany. E-mail: schmittel@chemie.uni-siegen.de
First published on 22nd April 2014
Abstract
A lab-on-a-molecule, based on the iridium(III)-imidazolium conjugate 1, allows competitive quantification of three distinct anions in a mixture using three interrogation channels: F− is detected and quantified via UV-Vis, H2PO4− using photoluminescence and AcO− using electrochemiluminescence.
The development of anion chemosensors is a burgeoning area of chemistry not only because of legal requirements for quantitative sensing in medicine and the environment but also due to vibrant academic challenges, such as molecular logic and dynamic receptors.1 Compared with cations, anions possess a lower charge-to-radius ratio, varying geometries and a distinctive pH dependence, so that anion detection is also affected by solvent properties.1a As a result, the design of selective anion receptors is much more demanding than that of cations, not to speak of multi-anion receptors. Among the concepts for multianalyte detection that are based on a single probe, such as lab-on-a-molecule,2 keypad lock,3 relay assay4 and displacement assay,1b the lab-on-a-molecule is known for its competitive assay. While the original conception of a lab-on-a-molecule was conceived with regard to molecular logic,2 we later extended the concept to multiplex detection in different channels.5 As such, it is an alternative strategy6 to array-based,7 highly integrated8 and combinatorial approaches.9 Without the need to integrate several binding/reaction sites, multi-channel lab-on-a-molecule probes ideally do not require complex synthesis but a specific binding mechanism and a unique signal response for each analyte within its detection channel. Although ample efforts have been devoted to and several examples have been addressed to,10 a lab-on-a-molecule for more than two anions has remained elusive so far.
Based on their unique photophysical properties, long excited-state lifetimes and stable redox states, ruthenium and cyclometalated iridium complexes display an armada of phenomena that are profitable for sensing, such as ligand-centred transitions (LC), interligand energy transfer (ILET) and/or metal–ligand charge transfer (MLCT).11 By using ligands with energy levels that are easily modulated by additives,12 we expected to trigger distinct photophysical changes for each analyte. Moreover, redox changes at the metal–ligand system should open further options to tune interactions with analytes, possibly causing unique electrochemiluminescence (ECL) responses.10e,h,i All these features and our recent results5,13 illustrate that ruthenium and iridium complexes are indeed great candidates for the lab-on-a-molecule approach.
Over the years, the imidazolium unit has been developed as an excellent receptor for anion binding,14 mostly based on electrostatic interactions and hydrogen bonding. However, the non-specificity of these interactions is a problem for selective sensing.1b We thus envisaged to implement anion-imidazolium selectivity by introducing specific auxiliary interactions and precise spatial arrangements.14 Our endeavours have led to the iridium-based lab-on-a-molecule 1 (Scheme 1), a molecular chemosensor for F−, H2PO4− and AcO−, whose energy levels are modulated depending on the interrogation channel and the anion–imidazolium interaction. The sensing of probe 1 was tested in three channels using the following array of anions: H2PO4−, HSO4−, F−, Cl−, Br−, PhCOO−, BF4−, PF6−, ClO4−, NO3−, CF3SO3−, MeSO3−, AcO− and TsO− (Fig. 1). Moreover, complex 2 was prepared for comparison to cross-examine the detection mechanism.
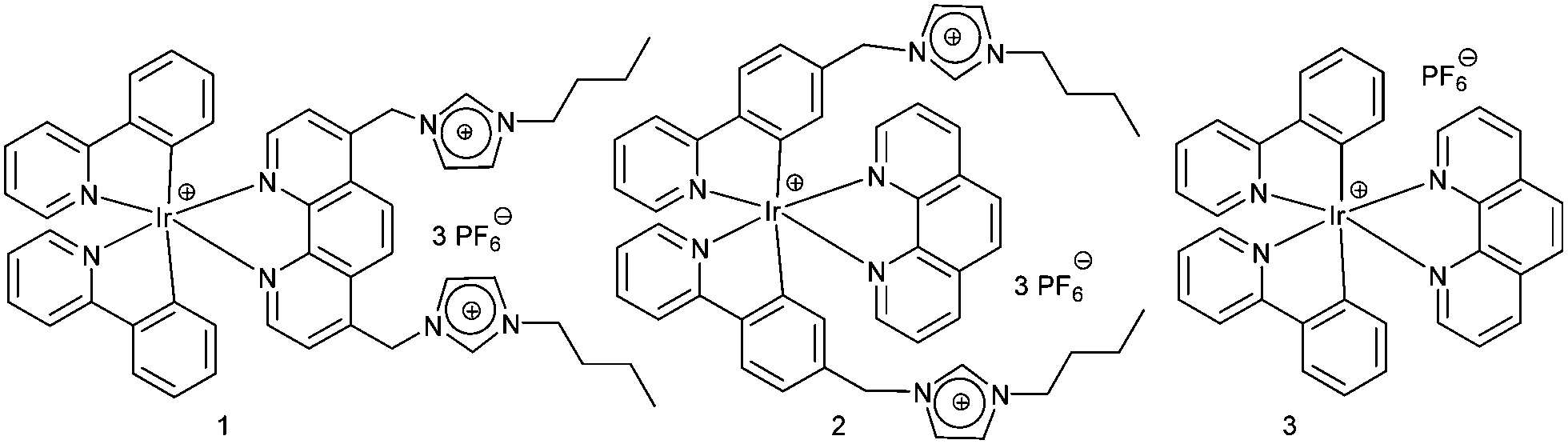 | ||
| Scheme 1 Structures of complexes 1–3. | ||
 | ||
| Fig. 1 Relative response of complex 1 in UV-Vis, PL (both upon addition of 5 equiv. of anions), and ECL (addition of 200 equiv. of anions). | ||
In the range of 350–500 nm, the UV-Vis absorption spectra of complexes 1 and 2 are very similar (Fig. S1, ESI†), while their response to anions is totally different. Upon addition of F− to a solution of 1 in acetonitrile (MeCN), a broad absorption and a hyperchromic effect at 457 nm were observed (Fig. 2). A less hyperchromic effect was seen upon addition of AcO−. Other anions did not induce any response in this range. In the case of probe 2, none of the anions caused any change at all.
 | ||
| Fig. 2 UV-Vis absorption spectra of probe 1 (10 μM) in the presence of F− (0 to 25 μM) in MeCN. Inset: titration of 1 with F− is monitored at 457 nm. | ||
The appearance of a new CT band at 457 nm for 1 in the presence of F− suggests that a new electron-donating unit is formed. Diagnostically, the 1H NMR of 1 in the presence of F− (2.8 equiv.) exhibits a downfield shift (Δδ = 0.62 ppm) for the imidazolium proton 2-H suggesting an ionic hydrogen-fluoride bond (Fig. S2, ESI†). In the presence of F− (2.8 equiv.), H/D exchange was observed in a mixture of CD3CN/CD3OD (v/v, 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at both 2-H and the methylene protons of complex 1. Conversely, only 2-H exchanged in complex 2 (Fig. S3 and S4, ESI†). Apparently, deprotonation of the methylene protons by F−/AcO− is the reason for an increased conjugation length in 1 (Scheme 2a).
:1) at both 2-H and the methylene protons of complex 1. Conversely, only 2-H exchanged in complex 2 (Fig. S3 and S4, ESI†). Apparently, deprotonation of the methylene protons by F−/AcO− is the reason for an increased conjugation length in 1 (Scheme 2a).
 | ||
| Scheme 2 Relevant interaction in the (a) UV-Vis and (b) ECL detection. | ||
The Job plot unveils a 1:2 stoichiometry for adduct 1·(F−)2 (Fig. S5, ESI†) and the UV-Vis titration indicates log β = 9.71 ± 0.30 as the association constant. A relationship (linear: 2–20 μM) allows quantification of F− at an LOD of 0.21 μM (limit of detection). In competition assays, the detection is only disturbed by the amphoteric acid–base pairs H2PO4− and HSO4− that will interact with any deprotonation scheme. Other anions do not interfere even in a competitive assay (Fig. S6, ESI†).
In the parent iridium complex 3, with the HOMO residing on the phenylpyridine and the LUMO on the phenanthroline ligand, the emission is centred at 597 nm.16 In contrast, 1 emits at 660 nm. With an oxidation potential at 1.41 V and three close reduction potentials at −0.99, −1.24 and −1.39 V vs. Fc*‡ (Fig. S7, ESI†), the band gap ΔE = Eox − Ered = 2.40 V of 1 is smaller than that (2.63 V) of complex 3 (Table 1). The lower energy emission of 1 indicates that its photoluminescence (PL) originates from low-lying triplet states of the auxiliary ligand (3LX) and ILET.17 In contrast, complex 2 exhibits a high-energy emission at 566 nm because the imidazolium unit, though not conjugated with the phenylpyridine ligand, lowers the HOMO energy while the LUMO energy is as in 3. Consequently, the band gap ΔE = 2.73 V of 2 is larger than that (2.63 V) of complex 3.
In the PL channel, both 1 and 2 exhibit a diagnostic light-up response toward H2PO4−. Upon addition of 5 equiv. of H2PO4− to 1, the emission increases 7-fold along with a blue shift from 660 to 607 nm (Fig. 3). In contrast, complex 2 displays an 8 nm red shift and an 8-fold PL enhancement at 564 nm (Fig. S8 and S9, ESI†). Job plots indicate that each imidazolium interacts with one equiv. of H2PO4− in both 1 (Fig. S10, ESI†) and 2 (Fig. S11, ESI†). UV-Vis (Fig. S12 and S13, ESI†) and PL titrations display comparable binding constants for both probes (logβ = 7.18 ± 0.10 for 1; logβ = 7.89 ± 0.10 for 2). A good relationship (linear: 6–34 μM) and LODs for 1 (68 nM) and 2 (65 nM) were obtained. In a competitive assay, only F− disturbs the detection by reducing the PL of 1 & H2PO4− at 607 nm by 64% (Fig. S15, ESI†) and that of 2 & H2PO4− at 564 nm by 30% (Fig. S16, ESI†), but this interference is fully eliminated using Me3SiCl as a trapping agent for F− (Fig. S17 and S18, ESI†). The presence of H2PO4− leads to a slight 1H NMR downfield shift at the imidazolium proton 2-H in 1 (Fig. S19, ESI†) and 2 (Fig. S20, ESI†), exclusively. The PL data were not disturbed by up to 5% (v/v) water in MeCN.
 | ||
| Fig. 3 PL spectra (λex = 272 nm) of 1 (10 μM) in the presence of H2PO4− (0 to 42 μM) in MeCN. Inset: PL titration (λem = 607 nm) of 1 with H2PO4−. | ||
The PL of iridium complexes is governed by the energy level of the lowest-lying ligand (triplet)11c and the energy gap law.12 Emission of 1 is weak due to the small energy gap between 3LX (imidazolium = im) and the ground state. Association with H2PO4− raises the LUMO level of 3LX(im) above that of the phenanthroline. As a result, the higher-energy 3MIrPpyLphenCT emission becomes favoured (Scheme 3). In contrast, the imidazoliums in 2 are attached to the phenylpyridines, the latter representing the HOMO. Upon addition of H2PO4−, all potentials of 2 shift cathodically except that of the phenanthroline ligand (Fig. S22, ESI†). Due to increasing orbital energies and a constant LUMO level, the energy difference between 1MLCT and 3LC states is reduced, which is well known to increase the quantum yield.12
 | ||
| Scheme 3 Energy diagram for the PL (a) of 13+, (b) of 13+ + H2PO4−, and (c) for the ECL of 13+. | ||
In oxidative ECL scans using tri-n-propylamine (TPrA) as a co-reactant (for mechanism, see ESI†), complex 1 displays an emission at 605 nm in MeCN-Tris buffered aqueous solution (9/1, v/v MeCN/10 mM Tris-ClO4 buffer, pH = 7.2). The notable difference Δλem = 55 nm between ECL and PL (λem = 660 nm) is readily explained by different HOMO/LUMO states in PL and ECL. The PL originates from an imidazolium-based LUMO (Epc = −0.99 V), and thus is not typical for [Ir(ppy)2(phen)]+ systems, while under ECL conditions, both the Ir4+-based SOMO and the phenanthroline-based LUMO are lowered in energy. As the latter falls below the energy level of the imidazolium unit (Scheme 3c), the ECL arises from a characteristic MIrPpyLphenCT that is typically located at 550–600 nm. In contrast, complex 2 displays an ECL emission centred at 563 nm, which is congruent with that observed in PL. In both excitation modes, the LUMO of 2 resides on the phenanthroline (Fig. S22, ESI†).
The difference in the ECL signature of both probes should have a bearing on anion detection. Indeed, probe 1 shows a highly selective ECL enhancement in the presence of AcO− (Fig. 4) with the response not being disturbed by any other anion from our set (Fig. S23, ESI†). A relationship (linear: 0.0–0.6 mM) allows quantification with an LOD = 0.17 mM. In contrast, complex 2 does not respond to AcO− at all in the ECL channel.
 | ||
| Fig. 4 ECL spectra of complex 1 (10 μM) in the presence of AcO− (0 to 2 mM) in MeCN-Tris buffer aqueous solution (9/1, v/v, 10 mM, pH = 7.2). Inset: titration (ECL at 605 nm) curve of 1 in the presence of AcO−. | ||
The selective ECL response of complex 1 to AcO− remains somewhat speculative. In a mechanistic test, propionate equally triggered ECL enhancement, while benzoic acid did not have any effect. We suggest that the positively charged imidazolium units and the iridium centre of 1+ form a tricationic cavity that serves to bind a single AcO− in a bifurcated hydrogen bond, a coordination mode that is well documented in spatially apt bisimidazolium salts.18 As the cavity (Scheme 2b) demands a small-sized anion, propionate still works, but benzoate fails. The binding of the acetate anion next to the phenanthroline raises the LUMO level (phenanthroline) of 1 slightly, shifting the emission moderately to higher energy (by 7 nm). Moreover, the ECL intensity is enhanced because the acetate serves to precoordinate TPrA+˙, which is the source of TPrA˙ (−1.83 V vs. Fc*),19 thus improving the injection of an electron into the LUMO.
According to this mechanistic hypothesis, probe 2 should indeed not experience any change in the ECL: (1) there is no small triply-charged cavity, and (2) due to acetate binding to the remote imidazolium units, the acetate is not brought next to the phenanthroline as a LUMO component.
Testing a sample containing all three anions in MeCN ([F−] = [H2PO4−] = 10 μM, [AcO−] = 0.20 mM), the following analysis was obtained using probe 1 (10 μM) and its single-anion calibration curves. The concentration of F− was determined to be 10.9 μM from the absorbance at 457 nm in the UV-Vis channel, the concentration of H2PO4− as 8.2 μM by using the PL intensity at 607 nm after adding Me3SiCl (10 μM) and the concentration of AcO− as 0.18 mM by the ECL intensity at 602 nm.
In summary, the first lab-on-a-molecule for quantifying three anions was established based on the iridium–imidazolium conjugate 1, despite the common notion that interactions between anions and imidazolium are rather unspecific. The interferences visible in competitive single-channel assays (Fig. S6, S15 and S23, ESI†) of 1 are eradicated using the supremacy of triple-channel detection. However, changes in the pH are not tolerated (ESI†).
Notes and references
- (a) G. T. Spence and P. D. Beer, Acc. Chem. Res., 2013, 46, 571 CrossRef CAS PubMed; (b) L. E. Santos-Figueroa, M. E. Moragues, E. Climent, A. Agostini, R. Martínez-Máñez and F. Sancenón, Chem. Soc. Rev., 2013, 42, 3489 RSC.
- (a) D. C. Magri, G. J. Brown, G. D. McClean and A. P. de Silva, J. Am. Chem. Soc., 2006, 128, 4950 CrossRef CAS PubMed; (b) A. P. de Silva and S. Uchiyama, Nat. Nanotechnol., 2007, 2, 399 CrossRef CAS PubMed; (c) A. P. de Silva, Chem. – Asian. J., 2011, 6, 750 CrossRef PubMed.
- D. Margulies, C. E. Felder, G. Melman and A. Shanzer, J. Am. Chem. Soc., 2007, 129, 347 CrossRef CAS PubMed.
- M. Dong, Y. Peng, Y.-M. Dong, N. Tang and Y.-W. Wang, Org. Lett., 2012, 14, 130 CrossRef CAS PubMed.
- M. Schmittel and H.-W. Lin, Angew. Chem., Int. Ed., 2007, 46, 893 CrossRef CAS PubMed.
- A. P. de Silva, Nature, 2007, 445, 718 CrossRef PubMed.
- (a) A. T. Wright and E. V. Anslyn, Chem. Soc. Rev., 2006, 35, 14 RSC; (b) D. R. Walt, Chem. Soc. Rev., 2010, 39, 38 RSC.
- O. A. Bozdemir, R. Guliyev, O. Buyukcakir, S. Selcuk, S. Kolemen, G. Gulseren, T. Nalbantoglu, H. Boyaci and E. U. Akkaya, J. Am. Chem. Soc., 2010, 132, 8029 CrossRef CAS PubMed.
- B. Rout, L. Unger, G. Armony, M. A. Iron and D. Margulies, Angew. Chem., Int. Ed., 2012, 51, 12477 CrossRef CAS PubMed.
- (a) N. Kaur and S. Kumar, Chem. Commun., 2007, 3069 RSC; (b) N. Singh, R. C. Mulrooney, N. Kaur and J. F. Callan, Chem. Commun., 2008, 4900 RSC; (c) N. Kaur and S. Kumar, Tetrahedron Lett., 2008, 49, 5067 CrossRef CAS PubMed; (d) N. Singh, N. Kaur, C. N. Choitir and J. F. Callan, Tetrahedron Lett., 2009, 50, 4201 CrossRef CAS PubMed; (e) E. Berni, I. Gosse, D. Badocco, P. Pastore, N. Sojic and S. Pinet, Chem. – Eur. J., 2009, 15, 5145 CrossRef CAS PubMed; (f) D. Y. Lee, N. Singh and D. O. Jang, Tetrahedron Lett., 2010, 51, 1103 CrossRef CAS PubMed; (g) D. Y. Lee, N. Singh and D. O. Jang, Tetrahedron Lett., 2011, 52, 3886 CrossRef CAS PubMed; (h) M. Schmittel and S. Qinghai, Chem. Commun., 2012, 48, 2707 RSC; (i) Q. Shu, L. Birlenbach and M. Schmittel, Inorg. Chem., 2012, 51, 13123 CrossRef CAS PubMed; (j) V. K. Bhardwaj, P. Saluja, G. Hundal, M. S. Hundal, N. Singh and D. O. Jang, Tetrahedron, 2013, 69, 1606 CrossRef CAS PubMed.
- (a) A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS; (b) L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143 CrossRef CAS; (c) Y. You and W. Nam, Chem. Soc. Rev., 2012, 41, 7061 RSC.
- J. Li, P. I. Djurovich, B. D. Alleyne, M. Yousufuddin, N. N. Ho, J. C. Thomas, J. C. Peters, R. Bau and M. E. Thompson, Inorg. Chem., 2005, 44, 1713 CrossRef CAS PubMed.
- (a) K. Chen and M. Schmittel, Analyst, 2013, 138, 6742 RSC; (b) K. Chen, J. W. Bats and M. Schmittel, Inorg. Chem., 2013, 52, 12863 CrossRef CAS PubMed.
- (a) Z. Xu, S. K. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 1457 RSC; (b) N. Ahmed, B. Shirinfar, I. S. Youn, A. Bist, V. Suresh and K. S. Kim, Chem. Commun., 2012, 48, 2662 RSC; (c) D. Zhang, X. Jiang, H. Yang, Z. Su, E. Gao, A. Martinez and G. Gao, Chem. Commun., 2013, 49, 6149 RSC.
- H. Lin, M. E. Cinar and M. Schmittel, Dalton Trans., 2010, 39, 5130 RSC.
- C. Dragonetti, L. Falciola, P. Mussini, S. Righetto, D. Roberto, R. Ugo, A. Valore, F. De Angelis, S. Fantacci, A. Sgamellotti, M. Ramon and M. Muccini, Inorg. Chem., 2007, 46, 8533 CrossRef CAS PubMed.
- Y. You and S. Y. Park, J. Am. Chem. Soc., 2005, 127, 12438 CrossRef CAS PubMed.
- Q.-S. Lu, L. Dong, J. Zhang, J. Li, L. Jiang, Y. Huang, S. Qin, C.-W. Hu and X.-Q. Yu, Org. Lett., 2009, 11, 669 CrossRef CAS PubMed.
- R. Y. Lai and A. J. Bard, J. Phys. Chem. A, 2003, 107, 3335 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, Job plots, UV-vis, competitive assays, DPV. See DOI: 10.1039/c4cc01421a |
| ‡ Fc* is decamethylferrocene, EFc* = −0.13 V vs. SCE. |
| This journal is © The Royal Society of Chemistry 2014 |