 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn amphiphilic, catalytically active, vitamin B12 derivative†
M.
Giedyk
a,
S. N.
Fedosov
b and
D.
Gryko
*a
aInstitute of Organic Chemistry Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dorota.gryko@icho.edu.pl; Web: http://ww2.icho.edu.pl/Gryko_group/index.html Fax: +48 22 632 66 81
bDepartment of Engineering Science, Aarhus University, Denmark
First published on 26th February 2014
Abstract
We performed the reaction of vitamin B12 with N,N-dimethylformamide dimethyl acetal for primary amide activation, and added MeOH as a nucleophile, to afford cobalester, the first amphiphilic cobalamin derivative. The unique combination of redox properties and solubility represents an asset for its use as a catalyst in C–C bond forming reactions.
Among many biologically important metals, Co, in the form of vitamin B12 (cyano-cobalamin 1), plays a unique role.1,2 B12 coenzymes are indispensable for catalyzing enzymatic rearrangements (coenzyme B12, adenosylcobalamin) and methylation reactions (methylcobalamin).1 The special ability of vitamin B12 (1) and other corrinoids to form a Co–C bond, combined with its facility in furnishing alkyl radicals via homolysis, has attracted the interest of many researchers, because corrinoids can be used as catalysts for C–C-bond forming reactions, which belong to the most challenging processes in organic synthesis.3 These catalytic reactions typically involve alkyl-cobalt complexes, which are formed in reactions of Co(I) species and an electrophile or a Co(II) and a radical.4 The most common chemical procedure utilizes the ‘supernucleophilicity’ of the Co(I) species (B12s) toward alkylating agents, such as alkyl halides. Homolysis of the Co–C bond generates a carbon-centered radical, which can, in situ, disproportionate, abstract H˙, or self-couple. In these reactions, B12 alkyl derivatives can be considered ‘reversible carriers’ of an alkyl radical.
To study the catalytic function of corrinoids in the hydrophobic microenvironment of enzyme active sites, Hisaeda and co-workers used cobyrinic acid derivatives as artificial enzymes. They were successfully used for dehalogenation, rearrangement, and ring expansion reactions.4–7 These corrinoids, devoid of the nucleotide loop, possess chemical and physical properties different from those of the parent vitamin B12. Importantly, in alkylcobalamins, the nucleotide enhances the ease of abstraction of the cobalt-alkyl group by an electrophile.8
We hypothesized that a hydrophobic derivative that retained the nucleotide loop would provide a better model for such haloenzymes and serve as better catalysts for organic reactions. This B12 derivative would require the conversion of the primary amide groups of cobalamin into esters. Typically, transformation of primary carboxamides into esters entails harsh reaction conditions. Brocchetta9 and Myers,10 however, showed that primary amides can be selectively activated and transformed into esters or secondary amides under mild conditions via the formation of formamidine, which subsequently reacts with nucleophiles (Table 1). Because the secondary amides remained intact, we reasoned that this methodology might be promising for selective modification of the primary amides in vitamin B12 leaving the nucleotide unaffected.
|
|
||||
|---|---|---|---|---|
| Entry | R | X | Substrate | Product |
| 1 |
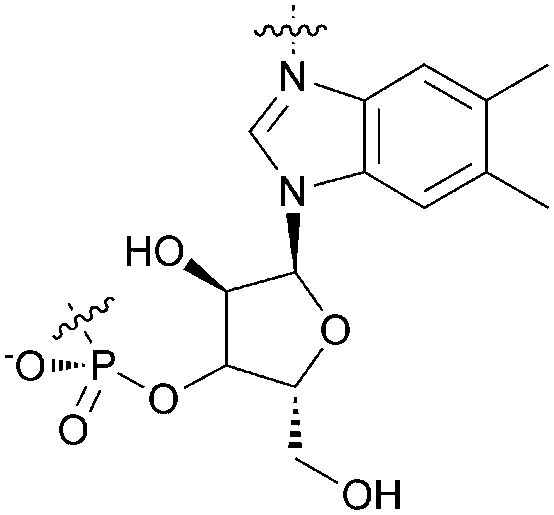
|
— | 1 | 3 |
| 2 | H | CN | 4 | 5 |
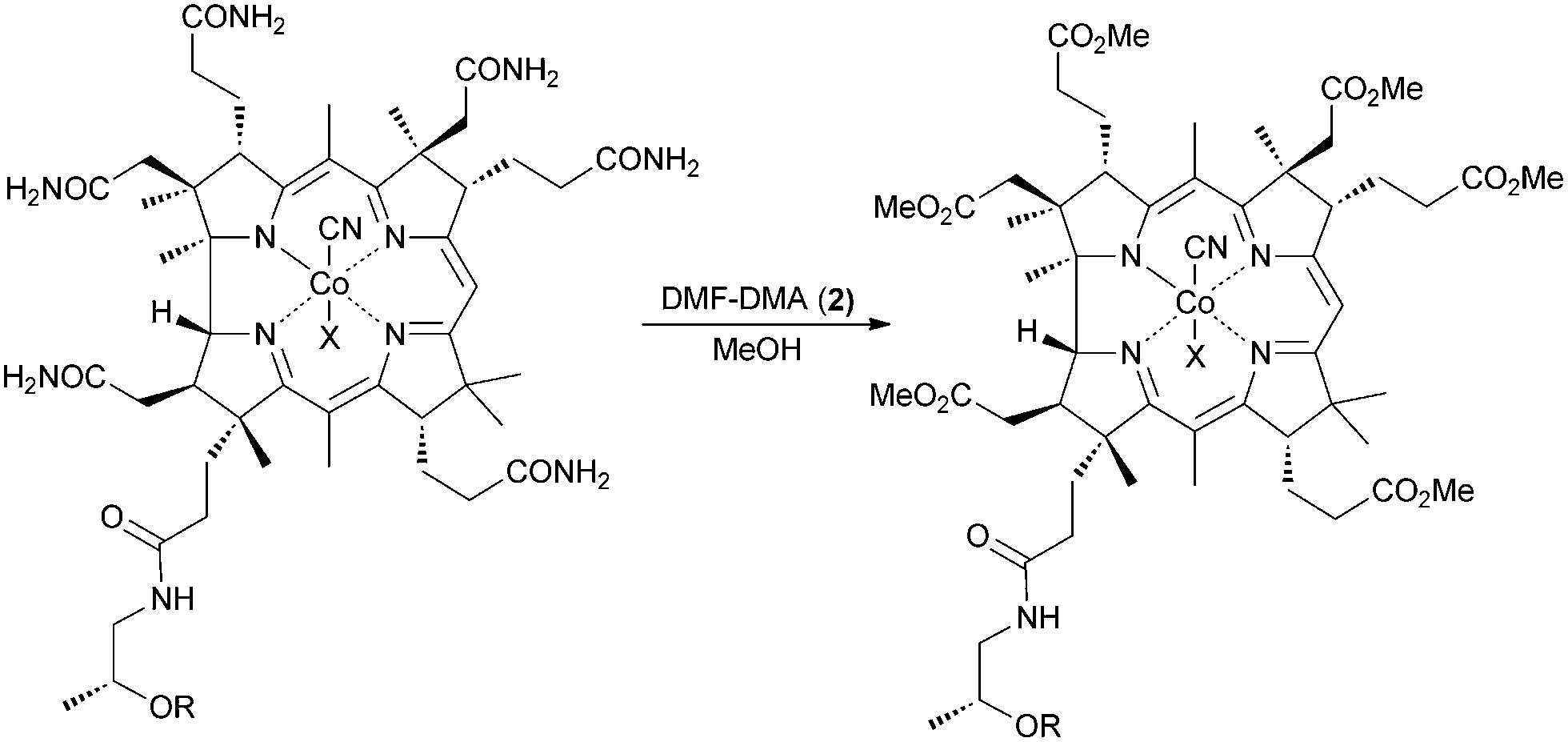
We report herein the synthesis of an amphiphilic, catalytically active, analogue of cobalamin (1), prepared via a one-step procedure, starting with vitamin B12 (1).
Initially, CNCbl (1) was treated with DMF-DMA (2) in MeOH at room temperature for up to 4 days. The reaction mainly led to pentamethyl ester with an intact nucleotide loop. Upon increasing the amount of the activating reagent and prolonging the reaction time, the desired cobalester (3) was obtained in very low amounts (1%). A substantial improvement in yield and selectivity was achieved when HFIP (hexafluoroisopropanol) was used as a solvent; this afforded cobalester (3) in 72% yield (for detailed optimization studies, see the ESI†). The positive influence of this polar, low nucleophilic solvent was previously reported; a similar effect was recently observed in hypervalent iodine-catalyzed reactions.11–13 Our method was also applied successfully to the synthesis of cobinester (5), a hydrophobic derivative of cobinamide (4).
The structure of cobalester (3) was confirmed by MS spectrometry, UV-Vis and NMR spectroscopy, and elemental analysis. The ESI-MS spectra showed a signal at m/z = 1445.57, which corresponded to a pseudomolecular ion [M+H]+. In the 1H NMR spectra, six proton resonances at 3.45, 3.68, 3.71, 3.72, 3.75, and 3.76 ppm, which were assigned to six –OMe groups, confirmed the replacement of six –CONH2 with –CO2Me groups. Ultimately, crystallization of the product by vapor diffusion of Et2O into EtOH gave crystals suitable for X-ray analysis, which unambiguously confirmed its structure (Fig. 1). As expected, product 3 displays UV-Vis characteristics that are nearly identical to the parent vitamin B12 (1), but significantly different from heptamethyl cobyrinate ((CN)2Cby(OMe)7) (see ESI†). The spectrum also confirmed that cobalester (3) exists in a ‘base on’ form.14 Derivative 3 did not bind to the specific B12-transporting proteins at concentrations as high as 1 μM, which confirms the crucial role of side chain amides in the B12 binding.
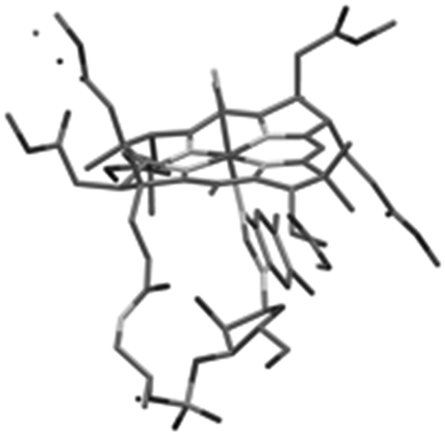 | ||
| Fig. 1 X-ray structure of cobalester (3). | ||
The corrinoid catalysis proceeds via the reduced forms, Co(II) and Co(I), whose formation depends on the nature of the axial ligands attached.1 In cobalamin and cobalester, these ligands are identical; consequently, their reduction potentials are very similar. Both compounds displayed the same cathodic Epc* values of −1.04 V vs. Ag/AgCl electrode (see ESI†), measured in an aqueous solution ([Tris] = 0.2 M, pH = 8.0) using CV.
With this redox behavior and excellent solubility in a broad range of organic solvents, the new amphiphilic ester 3 is capable of catalyzing C–C bond forming reactions. Reduced cobalamin can serve as a catalyst in various organic reactions, including dehalogenation,15 cyclopropane ring cleavage,16 additions to activated alkenes,15 functional group migrations,17 and alkene dimerization.18 Typical obstacles encountered in B12-catalyzed reactions include the low solubility of vitamin B12 (1) in organic solvents, high catalyst loading, and a limited number of reducing agents suitable for synthetic applications.
The radical homocoupling of benzyl bromide (6) is a typical model reaction for measuring the catalytic activity of reduced vitamin B12 (1) (Scheme 1). It was previously found that, depending on the Co-catalyst (vitamin B12, salen, CoPcTS), the reaction furnishes either toluene (7) or bibenzyl (8) via an anionic or a radical pathway, respectively.19
The benzyl radical formed can undergo further reactions, such as H radical abstraction from a solvent, homocoupling, or addition to activated double bonds.15,19,20 The cobalamin-catalyzed reaction predominantly afforded bibenzyl (8), but in a moderate yield (64%).
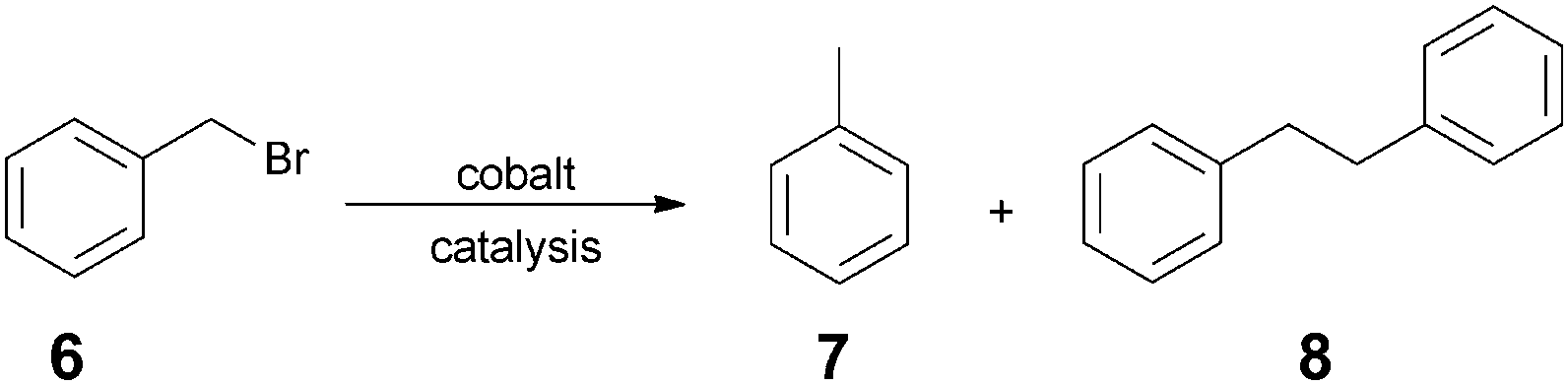 | ||
| Scheme 1 Homocoupling of benzyl bromide (4). | ||
Tanaka et al. showed that benzyl radicals can also be obtained by the photoinduced cleavage of cobalt(III) complexes, cis-[R2Co(bpy)2]ClO4, in the presence of benzyl halides.21 Other methods for obtaining this conversion include the homocoupling of halides in the presence of various metals22,23 (Ni, Mg, Mn, Zn, Cu, In, Cr, Fe, Ti), the homocoupling of Grignard compounds,24 oxidative coupling,25 Heck cross coupling,26 the Wittig reaction followed by reduction27etc. Though effective, these methods involve expensive reagents and/or ligands, high catalyst loading, environmental hazards, and difficulties in catalyst recycling. Consequently, interest remains high for the discovery of new, environmentally-benign methods that address these issues.
In our studies, the chemical reduction of cob(III)alester (3) to Co(I) species was performed using NaBH4/i-PrOH at an elevated temperature; then, it was reacted with benzyl bromide (6). In a typical experiment, all reagents were placed in a flask and allowed to react under deoxygenated conditions. The reaction at room temperature was very sluggish, but under microwave irradiation, it quickly proceeded to completion. A short optimization study demonstrated that the cobalester-catalyzed, microwave-assisted reaction of benzylbromide (6) at 90 °C for 15 min furnished the desired bibenzyl (8) in 84% yield (Table 2, entry 4). Gratifyingly, the catalyst loading could be as little as 0.5 mol%, which represents the lowest amount required to date in similar B12-catalyzed reactions. Surprisingly, when the reaction was heated in an oil bath, it furnished bibenzyl (8) in a low yield, despite the prolonged reaction time required for full conversion (entries 6, 7). For a detailed description of the optimization studies, see ESI.†
| Entrya | Catalyst loading [mol%] | Temperature [°C] | Time [min] | Yieldb [%] |
|---|---|---|---|---|
| a Reaction conditions: BnBr (0.25 mmol), i-PrOH (1 mL), NaBH4 (0.5 mmol), unless otherwise noted, the reactions were performed in a microwave reactor. b Isolated yields. c An oil bath was used instead of microwave irradiation. d Full conversion. | ||||
| 1 | 1.5 | 120 | 15 | 84 |
| 2 | 0.5 | 120 | 15 | 86 |
| 3 | 0.25 | 120 | 15 | 42 |
| 4 | 0.5 | 90 | 15 | 84 |
| 5 | 0.5 | 60 | 15 | 10 |
| 6c | 0.5 | Reflux | 15 | Traces |
| 7c | 0.5 | Reflux | 60 | 58d |
These reaction conditions render the method significantly simpler and more useful than other procedures reported to date, for example requiring electrochemical reduction, which limits their synthetic applicability.15,19–21,28 After establishing optimized reaction conditions, we investigated the scope of benzyl bromide (6) dimerization catalyzed by our cobalester (3). Benzyl bromides with either electron donating or electron withdrawing substituent were tested (Table 3).
| Entry | R | Catalyst loading [mol%] | T [°C] | Product | Yield [%] |
|---|---|---|---|---|---|
| a Reaction conditions: ArCH2Br (0.25 mmol), i-PrOH (1 mL), NaBH4 (0.5 mmol), 15 min; all reactions were performed in a microwave reactor. | |||||
| 1 | H | 0.5 | 90 | 8 | 84 |
| 2 | 4-I | 0.5 | 90 | 9 | 74 |
| 3 | 3-I | 0.5 | 90 | 10 | 70 |
| 4 | 4-NO2 | 0.5 | 90 | 11 | Traces |
| 5 | 4-NO2 | 2.5 | 120 | 11 | 56 |
| 6 | 2-CN | 2.5 | 120 | 12 | 52 |
| 7 | 4-F | 0.5 | 90 | 13 | 90 |
| 8 | 3-OMe | 0.5 | 90 | 14 | 91 |
All reactions with cobalester (3) gave the desired products, with yields exceeding 50%. Benzyl bromides with electron withdrawing substituents initially formed toluene derivatives. However, an increase in the catalyst loading (to 2.5%) and temperature (to 120 °C) led to the formation of the desired products, 11 and 12 (entries 5 and 6). Although the range of substrates was not exhaustive, the reaction appeared to be quite general. It has been shown that intramolecular coordination of the nucleotide function labilizes the organometallic C–Co bond in alkylcobalamins, which facilitates subsequent reactions.2
With our series of vitamin B12 derivatives, with and without the 5,6-dimethylbenzimidazole moiety, we were in a position to verify this hypothesis (assuming formation of benzyl-Cbl as an intermediate of the reaction). We evaluated the effectiveness of catalysts in the dimerization of benzyl bromide (6) under optimized conditions. A comparison of reactions in the presence of reduced cobalester (3) and (CN)2Cob(OMe)7 indicated that the nucleotide moiety increased the yield of bibenzyl (Table 4, entries 3, 5). Interestingly, ester derivatives were by far more efficient than vitamin B12 (1) and cobinamide, presumably due to differences in solubility (Table 4).
| Entry | Catalyst | Conversion [%] | Yieldb [%] |
|---|---|---|---|
| a Reactions were conducted with BnBr (0.25 mmol), i-PrOH (1 mL), NaBH4 (0.5 mmol), catalyst (0.5 mol%), at 90 °C (microwave heating) for 15 min. b Isolated yields. | |||
| 1 | Vitamin B12 (1) | 38 | 15 |
| 2 | Cobinamide (4) | 43 | 21 |
| 3 | Cobalester (3) | 100 | 84 |
| 4 | Cobinester (5) | 100 | 60 |
| 5 | (CN)2Cob(OMe)7 | 100 | 62 |
In summary, we have developed a new, efficient method for preparing cobalester (3), an amphiphilic vitamin B12 derivative. This derivative has esters instead of six amide groups, and an intact nucleotide. The reaction of vitamin B12 (1) with DMA-DMF (2) in HFIP provided the desired compound 3 in 72% yield.
As expected, cobalester (3) displayed UV-Vis spectra and oxidation–reduction characteristics similar to those of vitamin B12 (1). Both compounds differed significantly from the “incomplete” corrinoid heptamethyl cobyrinate, where the nucleotide moiety is absent, and instead, the second cyanide is coordinated. We confirmed the hypothesis that the intramolecular coordinating ligand plays a role in the radical reactions catalyzed by vitamin B12 derivatives. The microwave-assisted homocoupling of benzylbromide (6) catalyzed by cobalester (3) produced a higher yield when compared to reactions catalyzed by reduced (CN)2Cob(OMe)7. The amphiphilic character of this newly synthesized derivative (3) will facilitate a broader range of applications for vitamin B12-catalyzed reactions in organic synthesis.
We acknowledge the generous support from the Ministry of Science and Higher Education, grant no. 0145/DIA/2012/41.
Notes and references
- R. Banerjee, Chemistry and Biochemistry of B12, John Wiley & Sons, 1999 Search PubMed.
- B. Krautler, B. T. Golding and D. Arigoni, Vitamin B12 and B12-Proteins, WILEY-VCH, 2008 Search PubMed.
- R. Scheffold, S. Abrecht, R. Orlinski, H.-R. Ruf, P. Stamouli, O. Tinembart, L. Walder and C. Weymuth, Pure Appl. Chem., 1987, 59, 363–372 CrossRef CAS.
- Y. Hisaeda, T. Nishioka, Y. Inoue, K. Asada and T. Hayashi, Coord. Chem. Rev., 2000, 198, 21–37 CrossRef CAS.
- T. Ohno, A. Ogawa, Y. Hisaeda and Y. Murakami, J. Chem. Soc., Perkin Trans. 2, 1994, 2271 RSC.
- Y. Murakami, Y. Hisaeda and T. Ohno, Bull. Chem. Soc. Jpn., 1984, 57, 2091–2097 CrossRef CAS.
- M. A. Jabbar, H. Shimakoshi and Y. Hisaeda, Chem. Commun., 2007, 1653–1655 RSC.
- G. N. Schrauzer and J. H. Grate, J. Am. Chem. Soc., 1981, 103, 541–546 CrossRef CAS.
- P. L. Anelli, M. Brocchetta, D. Palano and M. Visigalli, Tetrahedron Lett., 1997, 38, 2367–2368 CrossRef CAS.
- T. A. Dineen, M. A. Zajac and A. G. Myers, J. Am. Chem. Soc., 2006, 128, 16406–16409 CrossRef CAS PubMed.
- A. Yoshimura, K. R. Middleton, M. W. Luedtke, C. Zhu and V. V. Zhdankin, J. Org. Chem., 2012, 77, 11399–11404 CrossRef CAS PubMed.
- K. Miyamoto, N. Tada and M. Ochiai, J. Am. Chem. Soc., 2007, 129, 2772–2773 CrossRef CAS PubMed.
- Y. Kita, H. Tohma, K. Hatanaka, T. Takada, S. Fujita, S. Mitoh, H. Sakurai and S. Oka, J. Am. Chem. Soc., 1994, 116, 3684–3691 CrossRef CAS.
- K. ó Proinsias, M. Giedyk and D. Gryko, Chem. Soc. Rev., 2013, 42, 6605–6619 RSC.
- J. Gao, J. F. Rusling and D. Zhou, J. Org. Chem., 1996, 61, 5972–5977 CrossRef CAS.
- H. Ogoshi, Y. Kikuchi, T. Yamaguchi, H. Toi and Y. Aoyama, Organometallics, 1987, 6, 2175–2178 CrossRef CAS.
- G. Pattenden, Chem. Soc. Rev., 1988, 17, 361–382 RSC.
- J. Shey, C. M. McGinley, K. M. McCauley, A. S. Dearth, B. T. Young and W. A. van der Donk, J. Org. Chem., 2002, 67, 837–846 CrossRef CAS PubMed.
- D.-L. Zhou, H. Carrero and J. F. Rusling, Langmuir, 1996, 12, 3067–3074 CrossRef CAS.
- J. F. Rusling and D.-L. Zhou, J. Electroanal. Chem., 1997, 439, 89–96 CrossRef CAS.
- S. Fukuzumi, K. Ishikawa and T. Tanaka, Organometallics, 1987, 6, 358–365 CrossRef CAS.
- J. Liu and B. Li, Synth. Commun., 2007, 37, 3273–3278 CrossRef CAS.
- C. D. Mboyi, S. Gaillard, M. D. Mabaye, N. Pannetier and J.-L. Renaud, Tetrahedron, 2013, 69, 4875–4882 CrossRef CAS PubMed.
- T. Nagano and T. Hayashi, Chem. Lett., 2005, 34, 1152–1153 CrossRef CAS.
- M. Blangetti, P. Fleming and D. F. O'Shea, J. Org. Chem., 2012, 77, 2870–2877 CrossRef CAS PubMed.
- M. L. Kantam, R. Chakravarti, V. R. Chintareddy, B. Sreedhar and S. Bhargava, Adv. Synth. Catal., 2008, 350, 2544–2550 CrossRef CAS.
- K. Hamza and J. Blum, Tetrahedron Lett., 2007, 48, 293–295 CrossRef CAS PubMed.
- K. S. Alleman and D. G. Peters, J. Electroanal. Chem., 1998, 451, 121–128 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, full characterisation of new compounds, biological tests and stability tests. CCDC 981457. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc01064g |
| This journal is © The Royal Society of Chemistry 2014 |