 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aminosilanetrithiol RSi(SH)3: an experimental and quantum-chemical study†
Yan
Li
ab,
Hongping
Zhu
*a,
Diego M.
Andrada
c,
Gernot
Frenking
*c and
Herbert W.
Roesky
*b
aState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, China. E-mail: hpzhu@xmu.edu.cn
bInstitut für Anorganische Chemie, Georg-August-Universität, Tammannstraβe 4, 37077-Göttingen, Germany. E-mail: hroesky@gwdg.de
cFachbereich Chemie, Philipps-Universität Marburg Hans-Meerweinstrasse, 35032-Marburg, Germany. E-mail: frenking@chemie.uni-marburg.de
First published on 10th March 2014
Abstract
An interesting aminosilanetrithiol RSi(SH)3 (R = N(SiMe3)-2,6-iPr2C6H3) has been prepared by the reaction of lithium aminosilanetrithiolate {RSi[SLi(THF)]3}2 with MeCOOH. Theoretical calculations indicate that the LP(N) → σ*(Si–S) and LP(S) → σ*(Si–S) electron donations remarkably contribute to the stabilization of the Si(SH)3 part of the molecule. RSi(SH)3 is the first example of a stable molecule containing three SH groups attached to one element.
Orthoformic acid (HC(OH)3) and its sulfur congener (HC(SH)3) are hypothetical molecules. Aqueous formic acid possibly contains HC(OH)3 which is considered to be extremely unstable.1 The analogous silicon species HSi(OH)3 and HSi(SH)3 have also been proposed and theoretically studied.2 RSi(OH)3 compounds with the bulky R substituents (R = alkyl, aryl, aryloxy, or aryl-substituted amide group or a metal cluster) have already been reported since the 1950s.3 They are extensively employed as building blocks for assembling lipophilic three-dimensional Si–O metal clusters.3b–d However, the sulfur analogue RSi(SH)3 has not been prepared so far.
It has been documented that some of the triorganosilanethiols (R3SiSH) and diorganosilanedithiols (R2Si(SH)2) can be obtained by a variety of methods including alcoholysis of SiS2,4 protonation of potassium silanethiolate,5 insertion of sulfur into triorganosilane,6 treatment of silylsulfide with hydrogen halide,7 and LiAlH4 reduction of a silanepolysulfide.8 More recently, a transition metal-trapped silylenylthiol LSi[Ni(CO)3]SH (L = HC[C(Me)CN-2,6-iPr2C6H3]2) has been achieved by 1,4-addition of H2S to L′Si[Ni(CO)3] (L′ = HC[C(Me)N-2,6-iPr2C6H3][C(CH2)N-2,6-iPr2C6H3]).9 In comparison with the preparation of the organosilanols,3 the synthesis of the organosilanethiols appears to be more complex. The approach to prepare organosilanetrithiol by reacting the R′SiCl3 (R′ = Me2iPrC or Me) precursor with Li2S10 or H2S/NEt3 in the presence of MeCl2SiSiCl2Me11 was not successful, and instead the silylsulfide clusters ((Me2iPrCSi)4S6 and (MeSi)4S5) were produced. An intermediate in these reactions might be the Si–S–M (M = Li, H) moiety, which further reacted to yield the Si–S–Si unit observed in the clusters.
RSi(OH)3 has been prepared by using RSiCl3 with a bulky R group which prevents the condensation shown in the previous work.3c Following the strategy of synthesizing RSi(OH)3 by controlled hydrolysis of RSiCl3 (1, R = N(SiMe3)-2,6-iPr2C6H3) in the presence of an amine as the HCl-acceptor,12 we used H2S instead of H2O. The experiments were carried out with different amines (NEt3, 2,6-iPr2C6H3NH2, or pyridine) at various temperatures.13 However, no reaction was observed.
Subsequently, we employed a salt metathesis reaction by treating 1 with Na2S or K2S, however, according to NMR analysis no transformation occurred. When we used a freshly prepared Li2S6 obtained from the reaction of sulfur with LiBEt3H, the reaction proceeded and several products were formed depending on the amount of Li2S and the reaction conditions. As illustrated in Scheme 1, the reaction of 1 with 5.3 equivalents of Li2S in THF was carried out at 25 °C and stirred for 10 days, affording {RSi[SLi(THF)]3}2 (2) as colorless crystals in high yield (85%). However, upon changing the temperature, time, and ratio of the precursors, the reaction only resulted in products RSiCl2SLi(THF)3 (3) and RSi[SLi(THF)](μ-S)2Si[SLi(THF)2]R (4). Compound 3 is an incompletely sulfurized species which can be isolated as colorless crystals in 80% yield, when the reaction was conducted under stirring at 10 °C for 24 h using three equivalents of Li2S. Increasing the reaction temperature to 35 °C and extending the reaction time to 72 h yielded a mixture of 3 and 4. The latter shows the formation of a Si(μ-S)2Si moiety, which is generated at a little higher temperature than 25 °C and is not observed during the formation of 2. Compared with the results reported in the literature,10,11 it is worth noting that a combination of the right RSiCl3,14 restricted temperature, long reaction time, and excess of Li2S is important to the successful and high yield production of 2.
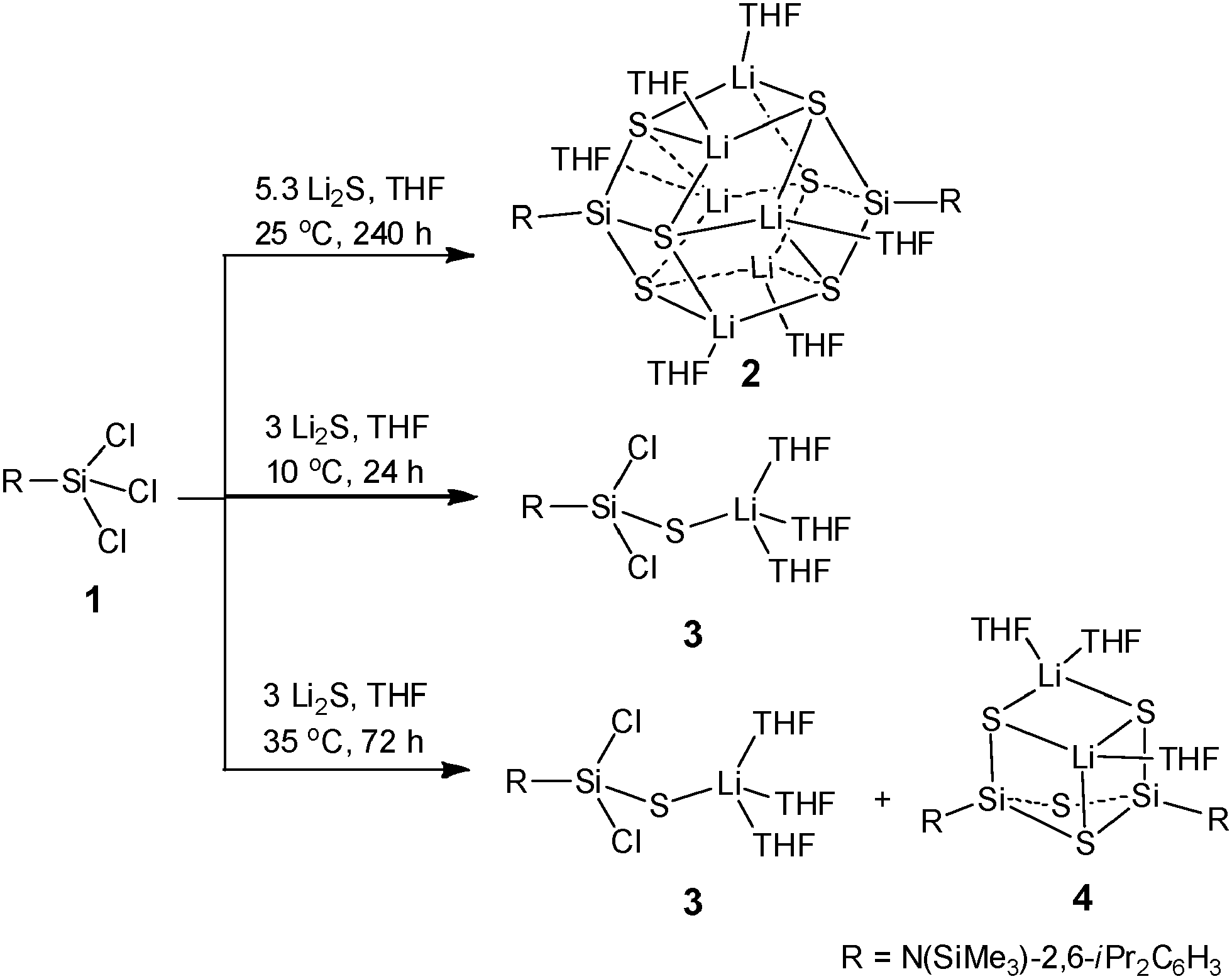 | ||
| Scheme 1 Reaction of 1 with freshly prepared Li2S under different reaction conditions to produce compounds 2–4. | ||
Compounds 2–4 are air and moisture sensitive and have been characterized by NMR spectroscopy and X-ray crystallography. Compound 2 shows symmetric patterns in 1H, 13C and 29Si NMR spectra in solution, indicating that all the R groups in 2 are equivalent. The CHMe2 resonance (4.05 ppm) of R in 2 is lowfield shifted when compared with those of 3 (3.60 ppm) and 4 (3.43 ppm). The 29Si NMR spectra of 2 display the respective resonances at δ 1.2 (SiMe3) and −4.2 ppm (SiS). The X-ray single-crystal structural analysis of 2 (Fig. 1) reveals that two Si(SLi)3 are assembled into a metal cage cluster which is comparable to those of the organosilanetriol-derived metallosiloxanes formed.3b–d The rhomboid-based dodecahedral Si2S6Li6 core of structure 2 contains six tetra-coordinate Li atoms each linked to one O atom from the THF molecule and three S atoms. Each of the six S atoms is located at the apex of a tetragonal pyramid and a SiLi3 four-membered ring forms the base of the pyramid. Similar structures containing Si2N6Li6,15 Si2P6Li6,16 and Ge2As6Li617 frameworks have been reported. The congener RSi(OX)3 (X = alkali metal) has not been prepared so far. The Si–S (2.1098(10)–2.1383(10) Å) and S–Li (2.398(5)–2.528(5) Å) bond lengths in 2 are comparable with those in 3 and 4, respectively. The X-ray structures and detailed descriptions of 3 and 4 are given in the ESI.†
 | ||
| Fig. 1 X-ray crystal structure of 2 with H atoms omitted for clarity. | ||
Compound 2 is a potential precursor for preparing aminosilanetrithiol. As expected, treatment of 2 with MeCOOH easily produced RSi(SH)3 (5) as colorless crystals in 40% yield (Scheme 2). The alternative use of CF3COOH, C6H5COOH, or p-MeC6H4SO3H is also possible but does not improve the yield.18 Compound 5 is air and moisture sensitive. It has a melting point of 154 °C, indicative of good thermal stability. In addition, 5 exhibits good solubility in common organic solvents such as n-hexane, toluene, diethyl ether, THF, and chlorinated hydrocarbons. The 1H NMR spectrum of 5 clearly shows the SH proton resonance at δ 1.18 ppm, which is comparable to those found in Tbt(Mes)Si(SH)2 and Tbt(Mes)Si(OH)SH (0.87–1.18 ppm).8 The SiS silicon resonance (2.7 ppm) is remarkably lowfield shifted even to a positive value in contrast to those of the SiO ones of the organosilanetriols (−40 to −90 ppm).3d,13 A strong SH absorption band is observed at ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 2543 cm−1 in the IR spectrum.
2543 cm−1 in the IR spectrum.
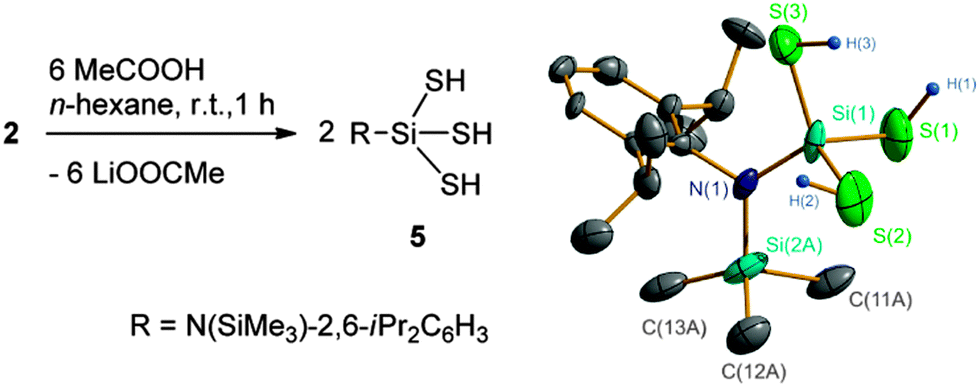 | ||
| Scheme 2 Synthesis and the crystal structure of 5. | ||
The structural analysis unambiguously reveals 5 to be a monomeric molecule (Scheme 2). However, the refinements indicate a severe disorder for the N-bonded SiMe3 and Si(SH)3 groups which are pseudo-symmetrically arranged along the N–Caryl axis, although the Me and SH groups show different appearances (see Fig. S4 in the ESI†). Treatment with a splitting mode results in a setting of the related bond lengths (Si–S, 2.10–2.25 ± 0.01; Si–C, 1.86 ± 0.01; SiSiMe3–N, 1.76 ± 0.01; SiSiS–N, 1.72 ± 0.01; S–H, 1.35 ± 0.03 Å) on the basis of the reported coordinate data. The final refinement gives convergence with bond parameters (Si–S, 2.101(8)–2.110(7) Å; S–H, 1.31–1.34(3) Å; ∠Si–S–H, 94(4)–109(2)°) comparable to those found in Tbt(Mes)Si(SH)2, Tbt(Mes)Si(OH)SH,8 and LSi[Ni(CO)3]SH9 as well as in the predicted [Si(SH)3]+.2a
To further understand the bonding matrix of compound 5, we carried out quantum chemical calculations on the basis of density functional theory (DFT). By optimizing the geometry of the molecule 5 at the M06-2X/def2-TZVPP level,19 the general structure (Fig. S6, ESI†) calculated is basically in agreement with the results obtained by the X-ray structural analysis (Fig. S4, ESI†). The SiSi(SH)3–N distance (1.712 Å) is computed to be ca. 0.020 Å longer than the experimental value while the SiSiMe3–N one (1.774 Å) is 0.014 Å shorter. Both the experimental and theoretical values are significantly shorter than the Si–N standard value (1.87 Å).20 The experimental Si–S bond lengths range from 2.101(8) to 2.110(7) Å, which are a little shorter than the calculated distances (2.139–2.147 Å).
We also performed the calculation on the 1H and 29Si NMR spectroscopic data by means of DFT-GIAO calculations.21 Since the NMR measurements were carried out in solution (CDCl3), we optimized the geometry of 5 using the PCM (Polarized Continuum Model)22 method in which the effect by solvent was considered. Therefore, the geometry of 5 optimized at the PCM(CHCl3)-M06-2X/def2-TZVPP level22 is only slightly different from the one at the M06-2X/def2-TZVPP level. It is computed that the chemical shifts for the Si(SH)3 group are at δ 2.02 ppm for the proton and 5.97 ppm for the silicon atom. The data for the hydrogen and silicon in the SiMe3 moiety are at δ 0.35 and 11.25 ppm, respectively. These data generally fit to the experimental5,9 and the other related computational data.23 Furthermore, the computed IR spectrum exhibits a band for the SH groups at 2753 cm−1, which is a little higher than the experimental value.
A natural bond orbital (NBO) analysis24 was accomplished to investigate the bonding situation of 5. The NBO results identify two N–Si bonds which are strongly polarized toward nitrogen (84% for the N atom and 16% for the Si atoms). The partial charges calculated suggest that the nitrogen atom carries a negative charge (−1.38 e) in a large extent while the silicon atoms have strong positive charges, namely, +1.91 e (for SiMe3) and +1.46 e (for Si(SH)3). The Wiberg bond order values P for SiSiS–N and SiSiMe3–N bonds have been calculated at different levels of theory (Table S5, ESI†). The data point out that the former (P = 0.61–0.63) has a higher bond order than the latter (P = 0.56–0.58). The former one can be explained by hyperconjugation of the lone pair orbital of the nitrogen, which is calculated by using a second-order perturbation theory included in the NBO method. The LP(N) → σ*(Si–S) donation amounts to 17.8 kcal mol−1 while the LP(N) → σ*(Si–C) hyperconjugation is only 11.2 kcal mol−1 (Table S3, ESI†). Furthermore, application of the second-order perturbation theory revealed delocalization by three stabilizing two-electrons from the sulfur (LP(S)) to the anti-bonding orbital σ*(Si–S), giving hints of a high conjugation of the Si(SH)3 fragment. The computed associated energies (ΔE(2)) are 13.54, 12.81 and 11.67 kcal mol−1 for each of the LP(S) → σ*(Si–S) interactions.
In summary, we have successfully synthesized the aminosilanetrithiol RSi(SH)3 (5) through protonation of its precursor {RSi[SLi(THF)]3}2 (2) by MeCOOH. Precise control of the reaction conditions is crucial for the synthesis of 2, while varying the reaction conditions led to the products 3 and 4. DFT calculations of 5 confirm the experimental data. The corresponding NBO analysis shows that the LP(N) → σ*(Si–S) and LP(S) → σ*(Si–S) donations remarkably contribute to the stabilization of the Si(SH)3 fragment. Compound 5 shows a structure containing three SH groups attached to one element. The reactivity studies of 5 are now in progress.25
This work was supported by the 973 Program (2012CB821704), the National Nature Science Foundation of China (91027014), and the Innovative Research Team Program (IRT1036 and 20923004). Support of the Deutsche Forschungsgemeinschaft (DFG) is highly acknowledged.
Notes and references
- (a) S. Böhm, I. Senf, H.-D. Schädler and J. Kuthan, THEOCHEM, 1992, 253, 73–82 CrossRef; (b) S. Böhm, D. Antipova and J. Kuthan, Int. J. Quantum Chem., 1996, 60, 649–655 CrossRef; (c) R. Glaser, G. S.-C. Choy, G. S. Chen and H. Grützmacher, J. Am. Chem. Soc., 1996, 118, 11617–11628 CrossRef CAS.
- (a) C. M. Marchand, U. Pidun, G. Frenking and H. Grützmacher, J. Am. Chem. Soc., 1997, 119, 11078–11085 CrossRef CAS; (b) A. Herman and W. Wojnowski, Struct. Chem., 1992, 3, 239–244 CrossRef CAS.
- See selected examples: (a) L. J. Tyler, J. Am. Chem. Soc., 1955, 77, 770–771 CrossRef CAS; (b) R. Murugavel, V. Chandrasekhar and H. W. Roesky, Acc. Chem. Res., 1996, 29, 183–189 CrossRef CAS and references therein; (c) R. Murugavel, A. Voigt, M. G. Walawalkar and H. W. Roesky, Chem. Rev., 1996, 96, 2205–2236 CrossRef CAS PubMed; (d) V. Chandrasekhar, R. Boomishankar and S. Nagendran, Chem. Rev., 2004, 104, 5847–5910 CrossRef CAS PubMed.
- R. Piekos and W. Wojnowski, Z. Anorg. Allg. Chem., 1962, 318, 212–216 CrossRef CAS.
- (a) J. E. Drake, B. M. Glavinčevski and R. T. Hemmings, Can. J. Chem., 1980, 58, 2161–2166 CrossRef CAS; (b) J. Pikies and W. Wojnowski, J. Organomet. Chem., 1989, 373, 317–326 CrossRef.
- H. Lange and U. Herzog, J. Organomet. Chem., 2002, 660, 36–42 CrossRef CAS.
- H. Beckers and H. Bürger, Z. Anorg. Allg. Chem., 2001, 627, 1217–1224 CrossRef CAS.
- (a) T. Tanabe, N. Takeda and N. Tokitoh, Eur. J. Inorg. Chem., 2007, 1225–1228 CrossRef CAS; (b) T. Tanabe, Y. Mizuhata, N. Takeda and N. Tokitoh, J. Organomet. Chem., 2009, 694, 353–365 CrossRef CAS.
- A. Meltzer, S. Inoue, C. Präsang and M. Driess, J. Am. Chem. Soc., 2010, 132, 3038–3046 CrossRef CAS PubMed.
- M. Unno, Y. Kawai, H. Shioyama and H. Matsumoto, Organometallics, 1997, 16, 4428–4434 CrossRef CAS.
- U. Herzog and G. Rheinwald, J. Organomet. Chem., 2001, 628, 133–143 CrossRef CAS.
- R. Murugavel, V. Chandrasekhar, A. Voigt, H. W. Roesky, H.-G. Schmidt and M. Noltemeyer, Organometallics, 1996, 14, 5298–5301 CrossRef.
- We have tried H2S in the liquid form at ca. −100 °C and allowed it to slowly warm to room temperature in the course of the reaction.
- We have also tested the group tolerance in the syntheses of compounds 2 and 5. Related results are shown in our recent patent which is still under authorization (CN10222598; Patent Application No. CN20111115911 20110505). However, compound 5 is selected as a typical example in this manuscript for profound experimental and theoretical studies.
- D. J. Brauer, H. Bürger, G. R. Liewald and J. Wilke, J. Organomet. Chem., 1985, 287, 305–320 CrossRef CAS.
- (a) M. Driess, G. Huttner, N. Knopf, H. Pritzkow and L. Zsolnai, Angew. Chem., Int. Ed. Engl., 1995, 34, 316–318 ( Angew. Chem. , 1995 , 107 , 354–356 ) CrossRef CAS; (b) M. Driess, C. Monsé and K. Merz, Z. Anorg. Allg. Chem., 2001, 627, 1225–1230 CrossRef CAS; (c) M. Driess, Adv. Inorg. Chem., 2000, 50, 235–284 CrossRef CAS.
- L. Zsolnai, G. Huttner and M. Driess, Angew. Chem., Int. Ed. Engl., 1993, 32, 1439–1440 ( Angew. Chem. , 1993 , 105 , 1549–1551 ) CrossRef.
- We tried HCl or HCl·NEt3 as the proton source. As a result, the chlorination happened and ionic (RCl2SiS)−(HNEt3)+ was isolated.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS; (b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- (a) K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS; (b) M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839–1862 CrossRef CAS PubMed.
- (a) S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef; (b) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS; (c) F. Jensen, J. Chem. Theory Comput., 2008, 4, 719–727 CrossRef CAS; (d) N. C. Handy and A. J. Cohen, Mol. Phys., 2001, 99, 403–412 CrossRef CAS; (e) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- (a) D. Cremer, L. Olsson and H. Ottosson, THEOCHEM, 1994, 313, 91–109 CrossRef; (b) L. Olsson, C.-H. Ottosson and D. Cremer, J. Am. Chem. Soc., 1995, 117, 7460–7479 CrossRef CAS.
- (a) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS; (b) A. E. Reed and F. Weinhold, J. Chem. Phys., 1985, 83, 1736–1740 CrossRef CAS; (c) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- Currently, we have obtained some preliminary results by isolating Ag4 clusters of composition [R(HS)2SiSAg]4 (6) and [R(HS)2SiSAg]2(AgMes)2 (7) from the reaction of 5 with (AgMes)4, which indicate a monofunctional reaction of the trithiol (5). Due to poor quality of the crystals of 7, a preliminary structure of this compound was determined. The synthesis of these two compounds and crystallographic data of 6 are included in ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and X-ray crystallographic and computational information. CCDC 973535 (2), 973536 (3), 973538 (4), 979368 (5), and 987309 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc00912f |
| This journal is © The Royal Society of Chemistry 2014 |