 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel P450-based biocatalyst for the selective production of chiral 2-alkanols†
Clemens J.
von Bühler
and
Vlada B.
Urlacher
*
Institute of Biochemistry, Heinrich-Heine University Düsseldorf, Universitätsstraße 1, 40225 Düsseldorf, Germany. E-mail: Vlada.Urlacher@uni-duesseldorf.de; Fax: +49 211 8113117; Tel: +49 211 8113866
First published on 25th February 2014
Abstract
A P450 monooxygenase from Nocardia farcinica (CYP154A8) catalyses the stereo- and regioselective hydroxylation of n-alkanes, still a challenging task in chemical catalysis. In a biphasic reaction system, the regioselectivity for the C2-position of C7–C9 alkanes was over 90%. The enzyme showed strict S-selectivity for all tested substrates, with enantiomeric excess (ee) of up to 91%.
Chiral linear alcohols are widely used as building blocks in the synthesis of pharmaceuticals, agrochemicals, pheromones and liquid crystals and therefore synthetic routes to these products are of high interest.1 Besides the difficult and less selective chemical routes,2 the enzymatic reduction of prochiral ketones catalysed by alcohol dehydrogenases is an established enzymatic process for the production of chiral alcohols.3 However, a prerequisite for this enzymatic approach is the availability of functionalised substrates, namely ketones. Therefore, an enzymatic approach for the production of chiral secondary alcohols via direct hydroxylation of inert alkanes by a monooxygenase seems to be more beneficial. Alkanes are cheap starting compounds that are easily produced from mineral oil and hence are available in high amounts. Enzyme-catalysed selective hydroxylation of alkanes comes along with the typical advantages of biocatalytic processes: no requirement for toxic metal catalysts or high reaction temperatures and only limited use of potentially toxic additional organic solvents.
Several enzyme groups have been described that are capable of this type of reaction.4 The number of oxygenases producing secondary and particularly 2-alcohols is however limited. The soluble methane monooxygenase (sMMO) and the particulate methane monooxygenase (pMMO) from Methylococcus capsulatus produce 2-alcohols from short chain alkanes but suffer from low expression.5 Furthermore fungal peroxygenases accept small and medium chain alkanes as substrates but display moderate regioselectivity.6 A very large and intensively studied group of enzymes catalysing hydroxylation reactions are the heme containing cytochrome P450 monooxygenases (P450s). Several wild type and engineered P450s are known to accept alkanes as substrates and especially CYP102A enzymes are known to produce 2-alcohols.7
Recently, we characterised two novel P450 enzymes belonging to the CYP154 family.8 CYP154A8 from Nocardia farcinica IFM 10152 proved to be quite regioselective. Thus, this enzyme was chosen for a detailed investigation of its suitability as a biocatalyst for n-alkane oxidation.
CYP154A8 was expressed in E. coli in soluble form and purified as reported earlier.8 A catalytically active P450 system was then reconstituted by the addition of the flavodoxin YkuN from Bacillus subtilis and E. coli flavodoxin reductase FdR.8 Reactions were performed in 500 μl reaction volume under NADPH cofactor regeneration by glucose dehydrogenase. The following alkanes were tested as substrates: heptane (C7), octane (C8), nonane (C9), decane (C10) and dodecane (C12). After extraction and derivatisation, the reaction mixtures were analysed via achiral and chiral GC coupled with MS. The stereoselectivity of the enzyme was chain length dependent. In comparison to authentic standards it could be determined that CYP154A8 showed strict S-selectivity for all tested alkanes: 84% ee for both 2-(S)-heptanol and 2-(S)-nonanol, 63% ee for 2-(S)-decanol and a maximum of 91% ee was found for 2-(S)-octanol (Fig. S1, ESI†). Dodecane was accepted by CYP154A8 but only trace amounts of products were formed and hence this alkane was not investigated further. In the case of decane oxidation, 27% 3-decanol was formed with 89% ee.
By monitoring the octane conversion over a period of 24 h it could be demonstrated that the ee value of the formed 2-(S)-octanol was not dependent on the degree of conversion, as is expected for an asymmetric synthesis (Fig. S2, ESI†).
So far the best-characterised P450s that catalyse this reaction with high stereoselectivity are the CYP102A1 mutants F87V/A328F,9 1-12G10 and 139-3,10,11 which all produce 2-octanol, but with significantly lower ee-values, i.e. 46% ee for 2-(R)-octanol, 39% ee for 2-(R)-octanol and 58% ee for 2-(S)-octanol, respectively. Thus CYP154A8 is the first P450 enzyme that is shown to produce highly enantiomerically enriched 2-(S)-octanol from octane.
However, substrate conversion in aqueous solution resulted in the formation of higher oxidation products, diols, which are produced upon exposure of the formed 2-alcohol to the enzyme as described earlier.8 In order to circumvent the formation of the undesired products, the reaction setup was modified: by employing the substrate as a second phase (20 vol% of the aqueous phase) a double benefit over the homogenous aqueous reaction system was achieved. First, the concentration of the substrate available to the enzyme was maximised, and second, the continuous extraction of formed products into the organic phase minimised or even prohibited the formation of higher oxidation products (Scheme 1). Partition coefficients for all 2-alcohols have been determined in the respective biphasic alkane–buffer-systems. Values ranged between log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PC7 1.0 ± 0.03 for 2-heptanol and logPC10 1.42 ± 0.1 (Table S1 and Section S4.4, ESI†). This altered reaction setup yielded 2-alcohols as main reaction products with over 80% selectivity. Considering that some of the 2-alcohol molecules were obviously further oxidised by CYP154A8 to the corresponding ketones, an overall preference towards the C2-position of up to 90% can be calculated (Fig. 1A–C and Fig. S3, ESI†).
PC7 1.0 ± 0.03 for 2-heptanol and logPC10 1.42 ± 0.1 (Table S1 and Section S4.4, ESI†). This altered reaction setup yielded 2-alcohols as main reaction products with over 80% selectivity. Considering that some of the 2-alcohol molecules were obviously further oxidised by CYP154A8 to the corresponding ketones, an overall preference towards the C2-position of up to 90% can be calculated (Fig. 1A–C and Fig. S3, ESI†).
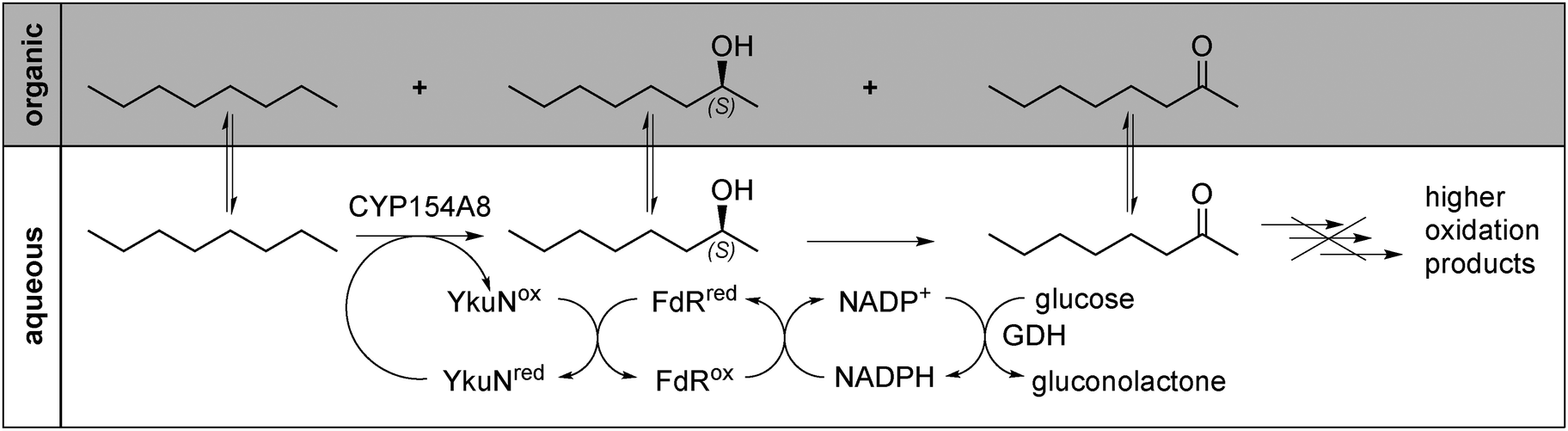 | ||
| Scheme 1 Hydroxylation of octane catalysed by CYP154A8 in a biphasic reaction system consisting of an organic substrate phase and an aqueous phase. The product scavenging effect of the organic phase prevents formation of higher oxidation products. YkuN (flavodoxin from B. subtilis), FdR (E. coli flavodoxin reductase), GDH (glucose dehydrogenase from B. megaterium). | ||
 | ||
| Fig. 1 Product distribution after hydroxylation of various alkanes: (A) heptane, (B) octane, (C) nonane, and (D) decane, determined from GC/MS peak areas and plotted as percentage of a specific product to the total amount of product (Fig. S3, ESI†). Where error bars cannot be seen, they are smaller than the linewidth. | ||
Similar to the stereoselectivity, the regioselectivity of CYP154A8 was chain length dependent and decreases for C10 down to 70% (Fig. 1D and Fig. S3, ESI†). For decane a significant amount of 3-decanol as well as its overoxidation product 3-decanone was detected. GC/MS data confirmed that 3-alkanones were not present for any other substrate but decane.
The benchmark for our reaction was the data published by Peters et al. (2003) for the CYP102A1 mutant 1-12G,11 and data for the CYP102A1 mutant F87V/A328F designed in our group.9 The mutant enzyme 1-12G produces 2-alcohols from alkanes ranging from C7–C10 with a corresponding selectivity of 76, 82, 86, and 86%. However, in all cases alcohols with a hydroxy group at the positions C1, C3 and C4 were observed as by-products. Compared to 1-12G, CYP154A8 showed a higher or similar regioselectivity depending on the chain length of the substrate, but its product distribution was limited to the C2- and partially C3-position only. The mutant F87V/A328F is very selective for the C2-position of octane (92%).9 Thus, the total regioselectivity of CYP154A8 for the C2-position was comparable to that of F87V/A328F.
In the next step challenges inherent to biphasic reaction systems such as interfacial surface area, low substrate solubility in the aqueous phase, and the choice of protein stabilising additives have been addressed. The optimised biphasic system included catalase to decompose H2O2, which might be formed by a shunt reaction in the catalytic cycle of P450s,12 BSA for protein stabilisation,13 ethanol as a cosolvent and the use of a rotator for mixing (Section S8 and Fig. S4, ESI†).
The following product titres were obtained after 24 h in the optimised biphasic reaction system: 3 mM 2-nonanol; 1.9 mM 2-heptanol; 2.2 mM 2-octanol; and 1.2 mM 2-decanol. Compared to values found for CYP102A1 mediated octane hydroxylation9,11 the total turnover numbers (TTNs) of CYP154A8 were much higher than those reported for F87V/A328F and in the same order of magnitude as those of 1-12G (Table 1). Moreover, these results demonstrated that even though the catalytic system consisted of five enzymes, this complexity apparently does not have a negative effect on product formation.
| Sub. | Product formation ratea,b | Couplingb (%) | Conc.c (mM) | TTNc,d | ee-(S)c (%) |
|---|---|---|---|---|---|
| a Given as nmol of 2-alcohol per nmol P450 per minute. b Determined using a 200 μl aqueous reaction system in multiwell plates with 1 mM substrate. c After 24 h in 500 μl of a biphasic reaction system. d Calculated as nmol of 2-alcohol per nmol P450. | |||||
| C7 | 1.13 ± 0.08 | 7.3 ± 0.9 | 1.9 | 2800 | 84 |
| C8 | 4.6 ± 1.04 | 21.1 ± 4.2 | 2.2 | 3200 | 91 |
| C9 | 3.73 ± 0.44 | 15. 9 ± 1.8 | 3.0 | 4400 | 84 |
| C10 | 0.37 ± 0.05 | 4.0 ± 0.6 | 1.2 | 1700 | 63 |
In P450 systems at various stages of the electron transfer chain, electrons can be diverted from a productive reaction, leading to unproductive cofactor consumption, which reduces the catalytic performance of the system. This can take place between redox partners as well as through the earlier mentioned shunt reactions in the P450 catalytic cycle. To evaluate the performance of the CYP154A8 system we calculated the coupling between NADPH consumption and product formation (Table 1 and ESI†). The rather low values (Table 1) are however in accordance with those reported for other artificial P450 redox chains oxidising non-natural substrates.14
At a closer look these data seem to be contradictory: the total turnover number (TTN) and the final product concentration were highest for nonane but its product formation rate and coupling were lower than for octane.
Since we attributed this discrepancy to the stability of the involved enzymes we designed experiments to verify our hypothesis: octane conversion was supplemented after 5.5 h with either additional glucose, glucose dehydrogenase (both for cofactor regeneration), YkuN/FdR or YkuN/FdR/P450. After a total reaction time of 24 h each case was compared to a standard reaction setup without this extra addition. Neither the addition of glucose nor that of glucose regenerating GDH resulted in an elevated final product concentration. As opposed to this, the supplementation of the reaction with the redox partners or the P450 led to about 15% more product which could even be further maximised (45%) by the simultaneous addition of YkuN, FdR and CYP154A8 (Fig. S5, ESI†). These results indicated that as suggested the stability of the redox partner proteins and that of CYP154A8 turned out to have a major influence on the degree of conversion. In the C7 to C12 alkane series the solubility decreases from 3.4 mg L−1 to 5.2 10−2 mg L−1.15 It is therefore reasonable to assume that enzyme denaturation is decreased with a longer alkane chain, due to reduced exposure. This in turn may explain the observed higher final concentrations of 2-nonanol as compared to 2-octanol (Table 1).
In conclusion, CYP154A8 from N. farcinica was identified as a novel regio- and stereoselective biocatalyst for the hydroxylation of inert n-alkanes. Depending on the substrate chain length, enzyme preference for oxidation at the C2-position was more than 90%. To the best of our knowledge, CYP154A8 is the only P450 monooxygenase where high regioselectivity for medium chain alkane hydroxylation coincides with a moderate to high enantiomeric excess of the formed 2-(S)-alcohols and high total turnover numbers. Furthermore, by means of reaction optimisation the total product amounts were significantly increased to levels that can compete with the best performing and well-established CYP102A1-based systems. But unlike those evolved CYP102A1 systems, CYP154A8 wild type shows already high selectivity, which might be even further increased by protein engineering methods. Compared to other enzymes like fungal peroxygenases both regio- and stereoselectivity of 2-alkanol production by CYP154A8 is significantly higher.6 This study demonstrates the potential of CYP154A8 in alkane oxidation. Simultaneously, the described biocatalyst implies additional optimisations and the transfer to a whole cell system which are currently under investigation.
Notes and references
- (a) K. Faber and M. C. Franssen, Trends Biotechnol., 1993, 11, 461 CrossRef CAS; (b) N. Öhrner, C. Orrenius, A. Mattson, T. Norin and K. Hult, Enzyme Microb. Technol., 1996, 19, 328 CrossRef; (c) R. A. Sheldon, J. Chem. Technol. Biotechnol., 1996, 67, 1 CrossRef CAS.
- R. H. Crabtree, J. Chem. Soc., Dalton Trans., 2001, 2437 RSC.
- W. Kroutil, H. Mang, K. Edegger and K. Faber, Curr. Opin. Chem. Biol., 2004, 8, 120 CrossRef CAS PubMed.
- (a) Y. Ji, G. Mao, Y. Wang and M. Bartlam, Front. Microbiol., 2013, 4, 58 Search PubMed; (b) J. B. Van Beilen and E. G. Funhoff, Appl. Microbiol. Biotechnol., 2007, 74, 13 CrossRef PubMed; (c) M. Bordeaux, A. Galarneau and J. Drone, Angew. Chem., Int. Ed., 2012, 51, 10712 CrossRef CAS PubMed.
- (a) S. J. Elliott, M. Zhu, L. Tso, H.-H. T. Nguyen, J. H.-K. Yip and S. I. Chan, J. Am. Chem. Soc., 1997, 119, 9949 CrossRef CAS; (b) S. I. Chan, K. H.-C. Chen, S. S.-F. Yu, C.-L. Chen and S. S.-J. Kuo, Biochemistry, 2004, 43, 4421 CrossRef CAS PubMed.
- S. Peter, M. Kinne, X. Wang, R. Ullrich, G. Kayser, J. T. Groves and M. Hofrichter, FEBS J., 2011, 278, 3667 CrossRef CAS PubMed.
- O. Lentz, V. Urlacher and R. D. Schmid, J. Biotechnol., 2004, 108, 41 CrossRef CAS PubMed.
- C. von Bühler, P. Le-Huu and V. B. Urlacher, ChemBioChem, 2013, 14, 2189 CrossRef PubMed.
- E. Weber, A. Seifert, M. Antonovici, C. Geinitz, J. Pleiss and V. B. Urlacher, Chem. Commun., 2010, 46, 944 Search PubMed.
- A. Glieder, E. T. Farinas and F. H. Arnold, Nat. Biotechnol., 2002, 20, 1135 CrossRef CAS PubMed.
- M. W. Peters, P. Meinhold, A. Glieder and F. H. Arnold, J. Am. Chem. Soc., 2003, 125, 13442 CrossRef CAS PubMed.
- P. R. Ortiz de Montellano, Cytochrome P450. Structure, mechanism, and biochemistry, Kluwer Academic/Plenum Publishers, New York, 3rd edn, 2005 Search PubMed.
- S. C. Maurer, K. Kühnel, L. A. Kaysser, S. Eiben, R. D. Schmid and V. B. Urlacher, Adv. Synth. Catal., 2005, 347, 1090 CrossRef.
- (a) A. Schallmey, G. den Besten, I. G. P. Teune, R. F. Kembaren and D. B. Janssen, Appl. Microbiol. Biotechnol., 2011, 89, 1475 CrossRef CAS PubMed; (b) M. Girhard, T. Klaus, Y. Khatri, R. Bernhardt and V. B. Urlacher, Appl. Microbiol. Biotechnol., 2010, 87, 595 CrossRef CAS PubMed; (c) S. G. Bell, C. F. Harford-Cross and L. L. Wong, Protein Eng., 2001, 14, 797 CrossRef CAS PubMed.
- National Library of Medicine (US), Division of Specialized Information Services, Hazardous Substances Data Bank, available at: http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?HSDB.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods. GC/MS chromatograms, time dependence of octane conversion, and supplementation experiments. See DOI: 10.1039/c4cc00647j |
| This journal is © The Royal Society of Chemistry 2014 |