 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Complexation-induced inversion of helicity by an organic guest in a dynamic molecular propeller based on a tristerephthalamide host with a two-layer structure†
Ryo
Katoono
*,
Kenshu
Fujiwara
and
Takanori
Suzuki
*
Department of Chemistry, Faculty of Science, Hokkaido University, Sapporo 060-0810, Japan. E-mail: katoono@sci.hokudai.ac.jp; tak@sci.hokudai.ac.jp; Fax: +81 11 706-2714; Tel: +81 11 706-3396
First published on 29th January 2014
Abstract
A tristerephthalamide host exhibited two helical geometries with (M)- and (P)-helicity, respectively, in terms of the twisting direction of a two-layer structure, and the helical preference switched upon complexation with a ditopic guest. In both uncomplexed and complexed states, the intramolecular transmission of chirality was responsible for the control of helicity.
A dynamic molecular propeller is an attractive motif for studies on transmission of chirality1,2 and inversion of helicity,3–7 since a propeller-shaped conformation is helically chiral and dynamically interconvertible to the antipodal conformer, similar to polymeric helices.8 A preference for a particular sense of dynamic helicity is a result of the intra-1 or intermolecular2 transmission of point chirality. When point chirality is transferred to dynamic helicity, two conformers with (P)- or (M)-helicity are no longer enantiomers, but rather are diastereomers. One of them is more stable and favored over the other due to the transmission of chirality. It is important that we understand how the helical preference switches in response to a change in the environmental conditions, such as the solvent,3 temperature,4 electric charge,5 or the addition of a guest.6,7 In the development of an asymmetric catalyst,3d,9 the ability to switch helical chirality enabled the selective production of either enantiomer from a single catalyst.3d Furthermore, it might be challenging for an artificial system to undergo a change in conformation from a helical form to other helical form(s) as well as inversion of helicity in response to external stimuli.
We used a dynamic molecular propeller to study the inversion of helicity accompanied by a change in conformation upon complexation with a guest, and designed tristerephthalamide hosts 1 with a two-layer structure (Fig. 1). This is different from the above-described simple switching of the helical preference of a diastereomeric pair through complexation with a guest.6 Instead, a helical molecule originally prefers a particular sense of (P)- or (M)-helicity due to the intramolecular transmission of chirality, and this preference changes in a newly generated helical structure upon complexation (complexation-induced change in conformation and inversion of helicity).7 Two planes of 1,3,5-triethynylbenzene arranged one above the other provide the potential for creating helical chirality in terms of the twisting direction (Scheme 1a).10 We denote the two conformers as uppercase P and M, even though they are enantiomers or diastereomers. To fix the two planes, we used a threefold terephthalamide. In a syn-form terephthalamide unit, disrotatory twisting of the two amide groups leads to a nonhelical conformation with a mirror plane in the unit, and conrotatory twisting allows the unit to adopt helical conformations that can undergo interconversion about the 2-fold axis of symmetry (Scheme 1b).11 We denote the two helical conformations in the terephthalamide unit as lowercase p and m, in addition to the global helicity of P/M. In the case where a nonhelical conformation (n) is energetically favored over helical forms (p/m), we can anticipate that threefold conformational switching from a nonhelical conformation (n) to a helical conformation (p/m) would result in the generation of propeller chirality in the whole molecule (ppp/mmm). This can be induced by supplying sufficient energy for transformation, e.g. by complexation with a hydrogen-bonding ditopic guest at the two amide carbonyls.11a,c,12 Helicity biasing would be enabled by the attachment of point chirality to the nitrogen atom(s) and/or by complexation with a chiral ditopic guest through the intra- or intermolecular transmission of point chirality to dynamic helicity (P/M and/or p/m).11,12 We envisioned that point chirality associated with a host could be intramolecularly transferred to dynamic helicity in different ways in response to an uncomplexed (Pnnn/Mnnn) or complexed state (Pppp/Mmmm). Thus, we attached point chirality (R) to an amide nitrogen as an internal chirality [(R,R,R)-1a] and used a chiral ditopic guest12 such as (R,R)-2 or (S,S)-2 to induce the host to transform (Fig. 1). In this communication, we report the successful design of a dynamic molecular propeller in which the helical sense was switched upon complexation (Scheme 2). The host preferred a particular sense of P or M due to the intramolecular transmission of point chirality in the absence of a guest, and this helical preference changed in a complexed state (inversion of helicity). During complexation, the host underwent conformational switching from a nonpropeller form (nnn) to a propeller form (ppp) (helicity generation and biasing).
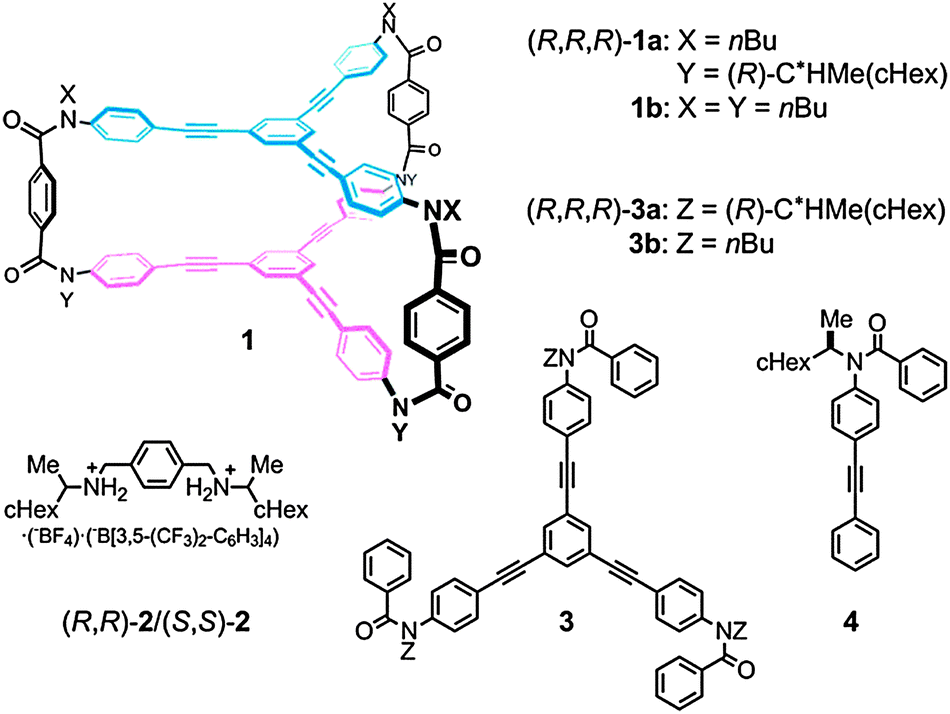 | ||
| Fig. 1 Chemical structures of hosts 1, chiral ditopic guests 2,12a and substructures 3 and 4. | ||
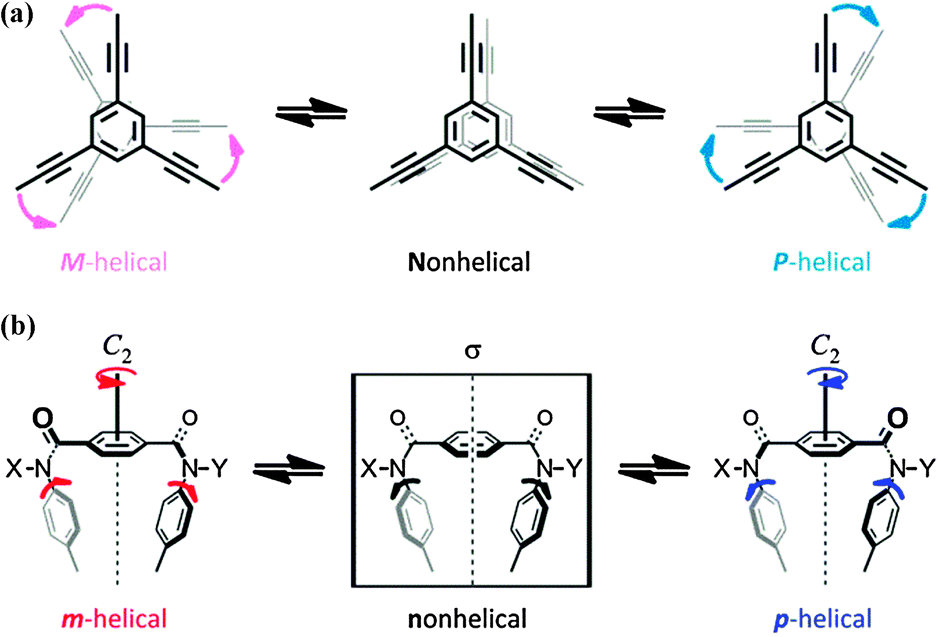 | ||
| Scheme 1 | ||
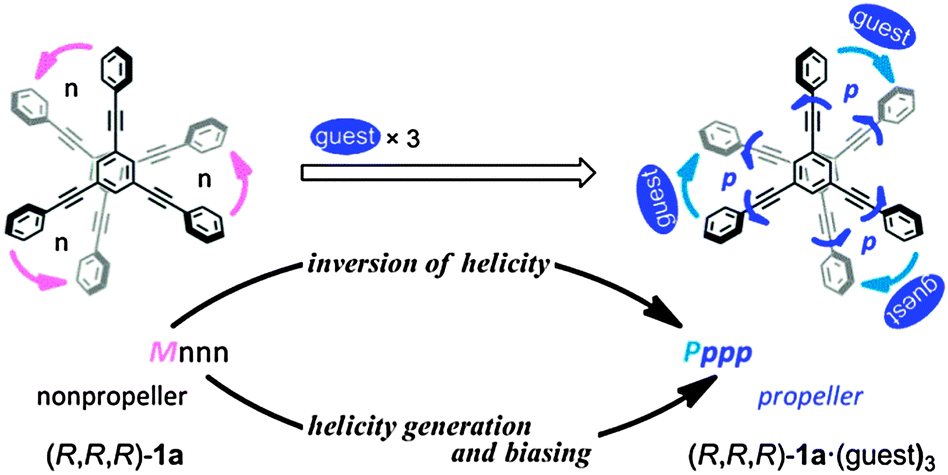 | ||
| Scheme 2 | ||
We prepared tristerephthalamide hosts 1a and b through a threefold condensation reaction of a trianiline and a tricarboxylic acid, both of which were derived from 1,3,5-triethynylbenzene13 (Scheme S1, ESI†). A conformational search for a model (R,R,R)-1a′ [X = Me, Y = (R)-C*HMe(cHex)] predicted several energy-minimized structures. The most energetically-minimized conformation was Mnnn (rel. 0 kJ mol−1) with two layers twisted in an (M)-helical manner, and each terephthalamide unit adopted a nonhelical form (Fig. 2a). A propeller arrangement was found for Pppp (+7.9 kJ mol−1) (Fig. 2b) as well as Mmmm (+14.2 kJ mol−1), and in both conformations, all six blades were twisted in a particular direction. It should be noted that Pppp was favored over Mmmm, since point chirality (R) preferred p-conformations with dynamic helicity in a terephthalamide unit. These predictions led us to expect that M-helicity would change to P-helicity when the host forms a complex with a ditopic guest at the two amide carbonyls to adopt a p-helical form (Scheme 2). The energy diagrams obtained for 1a′ and 1b′ are shown in Fig. S1 (ESI†).
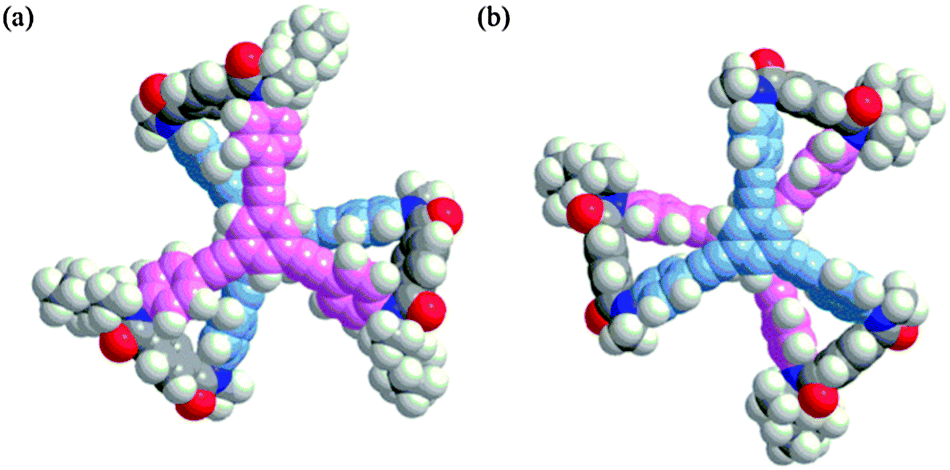 | ||
Fig. 2 Energy-minimized structures for a model (R,R,R)-1a′: (a) Mnnn (rel. 0 kJ mol−1) and (b) Pppp (+7.9 kJ mol−1), obtained by a conformational search using the MacroModel software (v9.9 Monte Carlo Multiple Minimum method, MMFF*, nonsolvated, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps). 000 steps). | ||
In the 1H NMR spectrum of (R,R,R)-1a, we observed a single set of averaged resonances assigned to C3 symmetry, which was maintained while the temperature was lowered to 223 K. During the VT measurements (223–293 K), we found a significant change in the chemical shift for the aromatic protons of a 1,3,5-triethynylbenzene unit (Fig. S2a, ESI†). We considered the change to be the result of the increased contribution of energetically higher conformers involved in the equilibrium at elevated temperatures, since a similar change was also observed for 1b (Fig. S2b, ESI†).
We first investigated the complexation of 1a with a ditopic guest 2 by monitoring complexation-induced changes in the chemical shift by 1H NMR spectroscopy, and spectra were measured in CDCl3 at ambient temperature. We confirmed that the host and guest formed a 1:3 complex by a significant upfield shift for both the phenylene protons of a terephthalamide unit in 1 and the phenylene protons in 2 (Fig. S3, ESI†). In addition, we found that the aromatic protons of a 1,3,5-triethynylbenzene unit were shifted downfield with an increase in guest equivalents (Fig. S3a, ESI†). This change indicated a conformational switching of 1a from the most stable conformation to some other conformation(s) and corresponded to the above-mentioned change with an increase in temperature (Fig. S2a, ESI†).
We then monitored the 1:3 complexation of (R,R,R)-1a with (R,R)-2 by CD spectroscopy, and spectra were measured in CH2Cl2 at 293 K. We found largely positive (Δε +19 at 310 nm) and bisignated Cotton effects (+19 at 280 nm and −22 at 258 nm) induced in the absorption region14 of 1a (Fig. 3a). These complexation-induced Cotton effects were totally different from the Cotton effects (−9.6 at 300 nm and −13 at 271 nm) obtained for (R,R,R)-1a itself through the intramolecular transmission of chirality, which appeared negatively throughout the whole region (Fig. 3a).16 We considered that this difference was the result of the conformational switching of (R,R,R)-1a upon complexation with (R,R)-2. The internal chirality was transmitted to newly generated dynamic helicity in a 1:3 complex, accompanied by a supramolecular transmission of guest chirality in a cooperative manner.17 The chiralities in 2 should have their own preference, as shown by the fact that small but similar Cotton effects were obtained by the complexation of 1b, which has no internal chirality, with (R,R)-2 through the supramolecular transmission of chirality to dynamic helicity that was generated upon complexation (Fig. 3b). We confirmed that a mirror image was induced by the addition of (S,S)-2. The conformational switching of (R,R,R)-1a upon complexation was also supported by the following experiments. When we gradually added the guest (R,R)-2 to a solution of (R,R,R)-1a, the complexation-induced Cotton effects and chemical shifts changed sigmoidally in CD and NMR spectra, respectively (Fig. S7, ESI†). During the 1:3 complexation, the guest bound to the host in an allosteric manner.11c,18
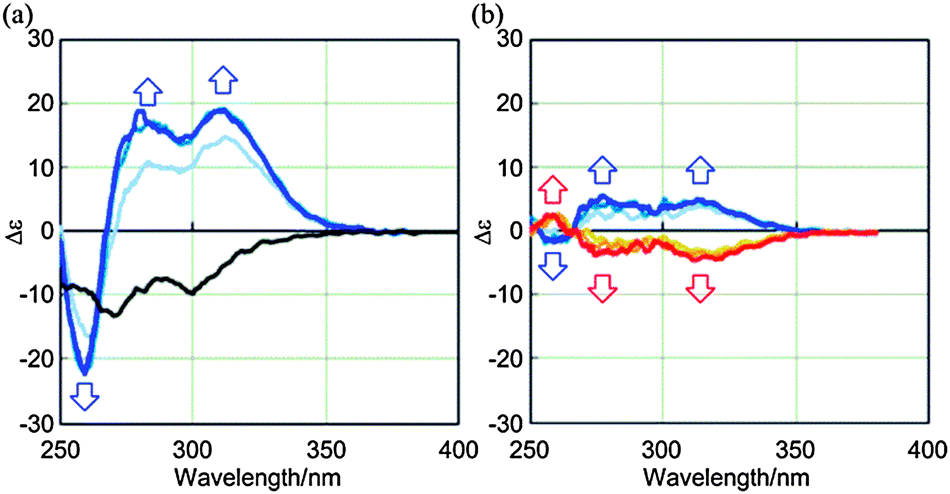 | ||
| Fig. 3 (a) Continuous changes in the CD spectrum of (R,R,R)-1a (8.4 × 10−5 M) upon complexation with a ditopic guest (R,R)-2 [0 (1a only, black line), 3, 6, and 12 equiv.]. (b) Continuous changes in the CD spectrum of 1b (8.8 × 10−5 M) upon complexation with a ditopic guest (R,R)-2 (blue lines) or (S,S)-2 (red lines) [3, 5, and 10 equiv.]. Molar CD values for 2 are <0.2 in the absorption region of 400–250 nm.12a All spectra were measured in CH2Cl2 at 293 K. | ||
In conclusion, we have demonstrated a complexation-induced inversion of helicity based on a dynamic molecular propeller. Two helical states with an inverse helical preference were required: the molecule prefers a particular sense of helicity in one state (Mnnn for an uncomplexed state), and this preference changes in the other state (Pppp for a complexed state). In both helical states, the point chirality (R) associated with the host was responsible for the control of helicity. Notably, we have presented a less well-developed motif for studies on the inversion of helicity upon complexation with an organic guest,6d,7b,c although metal ions6a and anions6b,c,7a,d,e have often been used as guests.
Notes and references
- J. Solá, S. P. Fletcher, A. Castellanos and J. Clayden, Angew. Chem., Int. Ed., 2010, 49, 6836 CrossRef CAS PubMed; D. Pijper and B. L. Feringa, Angew. Chem., Int. Ed., 2007, 46, 3693 CrossRef PubMed.
- C. Wolf and K. W. Bentley, Chem. Soc. Rev., 2013, 42, 5408 RSC; J. W. Canary, Z. Dai and S. Mortezaei, Compr. Chirality, 2012, 8(30), 600 Search PubMed.
- (a) H. Miyake, H. Sugimoto, H. Tamiaki and H. Tsukube, Chem. Commun., 2005, 4291 RSC; (b) J. Gregoliński, K. Ślepokura and J. Lisowski, Inorg. Chem., 2007, 46, 7923 CrossRef PubMed; (c) X. Jiang, Y.-K. Lim, B. J. Zhang, E. A. Opsitnick, M.-H. Baik and D. Lee, J. Am. Chem. Soc., 2008, 130, 16812 CrossRef CAS PubMed; (d) T. Yamamoto, T. Yamada, Y. Nagata and M. Suginome, J. Am. Chem. Soc., 2010, 132, 7899 CrossRef CAS PubMed; (e) N. Ousaka, Y. Takeyama and E. Yashima, Chem. Sci., 2012, 3, 466 RSC.
- J. Gregoliński and J. Lisowski, Angew. Chem., Int. Ed., 2006, 45, 6122 CrossRef CAS PubMed; H. Miyake, M. Ueda, S. Murota, H. Sugimoto and H. Tsukube, Chem. Commun., 2012, 48, 3721 RSC; H.-Z. Tang, P. D. Boyle and B. M. Novak, J. Am. Chem. Soc., 2005, 127, 2136 CrossRef PubMed; M. Fujiki, J. Am. Chem. Soc., 2000, 122, 3336 CrossRef.
- H. S. Barcena, A. E. Holmes, S. Zahn and J. W. Canary, Org. Lett., 2003, 5, 709 CrossRef CAS PubMed; S. Mortezaei, N. R. Catarineu and J. W. Canary, J. Am. Chem. Soc., 2012, 134, 8054 CrossRef PubMed.
- (a) S. Akine, S. Sairenji, T. Taniguchi and T. Nabeshima, J. Am. Chem. Soc., 2013, 135, 12948 CrossRef CAS PubMed; (b) J.-M. Suk, V. R. Naidu, X. Liu, M. S. Lah and K.-S. Jeong, J. Am. Chem. Soc., 2011, 133, 13938 CrossRef CAS PubMed; (c) R. M. Meudtner and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 4926 CrossRef CAS PubMed; (d) H. Kawamura, Y. Takeyama, M. Yamamoto, H. Kurihara, K. Morino and E. Yashima, Chirality, 2011, 23, E35 CrossRef CAS PubMed.
- (a) N. Ousaka, Y. Takeyama and E. Yashima, Chem.–Eur. J., 2013, 19, 4680 CrossRef CAS PubMed; (b) S. Akine, S. Hotate and T. Nabeshima, J. Am. Chem. Soc., 2011, 133, 13868 CrossRef CAS PubMed; (c) K. Maeda, H. Mochizuki, M. Watanabe and E. Yashima, J. Am. Chem. Soc., 2006, 128, 7639 CrossRef CAS PubMed; (d) H. Miyake, M. Hikita, M. Itazaki, H. Nakazawa, H. Sugimoto and H. Tsukube, Chem.–Eur. J., 2008, 14, 5393 CrossRef CAS PubMed; (e) S. Yano, M. Nakagoshi, A. Teratani, M. Kato, T. Onaka, M. Iida, T. Tanase, Y. Yamamoto, H. Uekusa and Y. Ohashi, Inorg. Chem., 1997, 36, 4187 CrossRef CAS.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102 CrossRef CAS PubMed.
- J. Lv, L. Zhang, Y. Zhou, Z. Nie, S. Luo and J.-P. Cheng, Angew. Chem., Int. Ed., 2011, 50, 6610 CrossRef CAS PubMed.
- Y. Rubin, T. C. Parker, S. J. Pastor, S. Jalisatgi, C. Boulle and C. L. Wilkins, Angew. Chem., Int. Ed., 1998, 37, 1226 CrossRef CAS; Y. Tobe, R. Umeda, M. Sonoda and T. Wakabayashi, Chem.–Eur. J., 2005, 11, 1603 CrossRef PubMed.
- (a) R. Katoono, H. Kawai, M. Ohkita, K. Fujiwara and T. Suzuki, Chem. Commun., 2013, 49, 10352 RSC; (b) R. Katoono, H. Kawai, K. Fujiwara and T. Suzuki, Chem. Commun., 2008, 4906 RSC; (c) R. Katoono, H. Kawai, K. Fujiwara and T. Suzuki, Chem. Commun., 2005, 5154 RSC; (d) R. Katoono, H. Kawai, K. Fujiwara and T. Suzuki, Tetrahedron Lett., 2004, 45, 8455 CrossRef CAS.
- (a) R. Katoono, H. Kawai, K. Fujiwara and T. Suzuki, J. Am. Chem. Soc., 2009, 131, 16896 CrossRef CAS PubMed; (b) R. Katoono, H. Kawai, K. Fujiwara and T. Suzuki, Tetrahedron Lett., 2006, 47, 1513 CrossRef CAS.
- E. Weber, M. Hecker, E. Koepp and W. Orlia, J. Chem. Soc., Perkin Trans. 2, 1988, 1251 RSC.
- The absorption maxima, measured in CH2Cl2 at ambient temperature, were 291sh. nm (logε 5.09) and 277 nm (5.13) for (R,R,R)-1a, 291 nm (logε 5.11) and 281 nm (5.11) for 1b, which were hypsochromically shifted compared to those for substructures (R,R,R)-3a [λmax 313 nm (logε 5.01), 305sh. (4.98)], 3b [313 nm (5.02), 305sh. (4.99)] (Fig. S4, ESI†) and the parent 1,3,5-tris(phenylethynyl)benzene15 [305 nm (4.93) in CHCl3].
- Y. Yamaguchi, T. Ochi, S. Miyamura, T. Tanaka, S. Kobayashi, T. Wakamiya, Y. Matsubara and Z. Yoshida, J. Am. Chem. Soc., 2006, 128, 4504 CrossRef CAS PubMed.
- Some of the Cotton effects at around 271 nm negatively increased (Δε −12 at 303 K to −18 at 263 K) with a decrease in temperature to 263 K (Fig. S5a, ESI†). This change may suggest that a pair of helical conformations, such as Mnnn and Pnnn, was in equilibrium, and the population of each component changed at lower temperatures. Even though a diastereomeric pair of two helical conformers with M or P helicity was in equilibrium, their chiroptical signals might not be very striking due to the nonhelical nature of a terephthalamide unit.11a–c The Cotton effects observed for (R,R,R)-1a had the same appearance as those for (R,R,R)-3a. The Cotton effects for (R,R,R)-3a were similar in shape and triple the strength of those for (R)-4 (Fig. S5b, ESI†).
- Similar but attenuated Cotton effects were induced when the antipodal ditopic guest (S,S)-2 was added to a solution of (R,R,R)-1a (Fig. S6, ESI†). In the complex, the internal chirality (R) was predominantly transmitted to newly-generated dynamic helicity in a competitive manner.
- T. Ikeda, K. Sada, S. Shinkai and M. Takeuchi, Supramol. Chem., 2011, 23, 59 CrossRef CAS and references therein.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, UV and CD spectroscopic data, and experimental details of preparation of a new compound. See DOI: 10.1039/c4cc00323c |
| This journal is © The Royal Society of Chemistry 2014 |