 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new metal–organic framework with ultra-high surface area†
Ronny
Grünker
a,
Volodymyr
Bon
a,
Philipp
Müller
a,
Ulrich
Stoeck
a,
Simon
Krause
a,
Uwe
Mueller
b,
Irena
Senkovska
a and
Stefan
Kaskel
*a
aDepartment of Inorganic Chemistry, Technische Universität Dresden, Bergstrasse 66, D-01069 Dresden, Germany. E-mail: stefan.kaskel@chemie.tu-dresden.de
bHelmholtz-Zentrum Berlin für Materialien und Energie, BESSY-MX Group, Albert-Einstein-Strasse 15, D-12489 Berlin, Germany
First published on 4th February 2014
Abstract
A new mesoporous MOF, Zn4O(bpdc)(btctb)4/3 (DUT-32), containing linear ditopic (bpdc2−; 4,4′-biphenylenedicarboxylic acid) and tritopic (btctb3−; 4,4′,4′′-[benzene-1,3,5-triyltris(carbonylimino)]tris-benzoate) linkers, was synthesised. The highly porous solid has a total pore volume of 3.16 cm3 g−1 and a specific BET surface area of 6411 m2 g−1, adding this compound to the top ten porous materials with the highest BET surface area.
During the last two decades metal–organic frameworks (MOFs) have continuously set new records in terms of specific surface area and pore volume, pushing the high pressure storage capacity for energetic molecules such as hydrogen and methane further and further.1
In order to avoid the disadvantageous formation of interpenetrated structures we propose the use of two different types of elongated linkers, one with a polar functionality, and another with a purely aromatic core structure. In the following we will show that this concept leads to a highly porous non-interpenetrated MOF structure but requires a distinct activation procedure due to the stronger interaction forces between solvating molecules and the polar functional groups of one linker.
As the polar tritopic linker 4,4′,4′′-[benzene-1,3,5-triyltris(carbonylimino)]trisbenzoic acid (H3btctb), which has been very successfully applied in the synthesis of porous MOFs,2 was used. 4,4′-Biphenylenedicarboxylic acid (H2bpdc) was chosen as the more hydrophobic linker because of the ideal di- and tritopic linker length ratio (LD/LT) of 0.63. According to Matzger et al., for the formation of copolymerization MOFs, the geometric factor describing the length ratio of the linkers as well as the connectivity to the cluster plays a key role in the formation of the given structure.3 For known MOFs the LD/LT ratios lie within a region from 0.44 to 0.66 (Table S1, ESI†).
The solvothermal reaction of H3btctb and H2bpdc with zinc nitrate yields coffin-shaped transparent crystals of Zn4O(bpdc)(btctb)4/3 (DUT-32, DUT – Dresden University of Technology), while an excess of H3btctb or H2bpdc in the reaction mixture leads to the formation of byproducts: Zn4O(btctb)2 (DUT-59), Zn4O(btctb)2(DMF)(H2O)1.5 (ref. 2a) or Zn4O(bpdc)3 (IRMOF-10).4 DUT-59 is isoreticular to MOF-1775 (for more details see ESI†).
Interestingly, DUT-32 has a framework isorecticular to UMCM-26 with a rare umt topology crystallizing in the P63/m space group.‡ The phase purity of bulk samples of DUT-32 was confirmed by powder X-ray diffraction measurements (see ESI†). The overall composition of the solvated material was determined by thermal (Fig. S7, ESI†) and elemental analysis, as well as 1H-NMR (Fig. S8, ESI†), and can be described as Zn4O(bpdc)(btctb)4/3(DEF)39.7(H2O)11.3.
The structure of DUT-32 consists of two symmetry-independent Zn4O6+ octahedral secondary building units. Both of them are surrounded by four btctb3− and two bpdc2− linkers. The ditopic bpdc2− ligands at the first Zn4O6+ cluster are arranged in a trans orientation, while the cis orientation of linkers relative to each other is found at the second cluster (Fig. 1 and Fig. S5, ESI†). With these types of connectivity, a 3D framework is constructed with three different but huge mesopores (30 × 40 Å, 28 × 32 Å and 20 × 26 Å, considering van der Waals radii of the atoms), and one smaller micropore (14 × 18 Å) (Fig. S1 and S2, ESI†). The crystallographic density of the desolvated DUT-32 is extremely low, only 0.27 g cm−3, and is close to the density of MOF-210 (0.25 g cm−3).7
 | ||
| Fig. 1 (a) Two crystallographically independent diastereomeric SBUs in DUT-32; (b) simplified representation of DUT-32. Cluster – green, tritopic linker – blue, ditopic linker – orange. | ||
However, not only the crystallographic porosity, but also real gas accessible volume of the compound generated after removal of solvent molecules are crucial for the manufacture of porous materials for numerous areas of application. Activation of DEF-containing DUT-32 samples by conventional heating under vacuum results in a non-porous materials. Similar results were observed for the solvent-exchange activation method using dichloromethane, chloroform or alcohols like methanol or ethanol.
For supercritical drying, the amide solvent incorporated into the pores was exchanged first with absolute ethanol, which shows high miscibility with liquid CO2 and is commonly used for MOF activation. The resulting material exhibits an unexpected type IV nitrogen physisorption isotherm and relatively low BET surface area (838 m2 g−1) and micropore volume (0.36 cm3 g−1) (Fig. S9, ESI†) indicating a network distortion. However, a very high total pore volume of 2.91 cm3 g−1 was calculated. PXRD measurements performed for the sample after drying indicate a complete loss of crystallinity.
Interestingly, a resolvation of this amorphous material with DEF and heating at 373 K for one day restores the crystallinity partly (Fig. S6a, ESI†). We suggest that due to its ability to form hydrogen bonds, ethanol binds quite strongly to the amide moieties of the network. Therefore, a complete exchange of the ethanol by CO2 within the pores does not occur, leading to a structural rearrangement with a loss of crystallographically defined porosity during removal of residual ethanol. A complete structural collapse of the network probably does not occur because of the extremely high total pore volume which is close to the theoretical value of 3.21 cm3 g−1, the completely intact morphology of the dried crystals and the recovery of the network by resolvation.
For the supercritical drying process the choice of the solvent is mainly based on its miscibility with liquid CO2.8,9 Therefore, the influence of solvents with minor polarity on the porosity was investigated. Applying the same drying protocol but using anhydrous acetone instead of ethanol on the same material (sample mass 100 mg) with a resting time in CO2 of one day (replacing the liquid CO2 3 times a day) resulted in a material with completely different textural properties. PXRD measurements show that the crystallinity of the sample remains intact (Fig. S6, ESI†). This is also reflected in the N2 physisorption isotherm which shows a typical shape for a MOF material with small mesopores and reaches saturation at 935 cm3 g−1, indicating a total pore volume of 1.44 cm3 g−1 (Fig. S9b, ESI†).
However, this value remains too low in comparison to the geometrically calculated pore volume of 3.21 cm3 g−1, indicating insufficient activation. Interestingly, upon decreasing the amount of the sample for SCD drying from 100 mg to 30 mg and keeping the resting time in liquid CO2 and replacement cycles the same (one day, three times a day, respectively), a sample with a significantly enhanced porosity was obtained. The adsorbed N2 volume reaches a value of 1470 cm3 g−1 representing a total pore volume of 2.27 cm3 g−1 (Fig. S9b, ESI,† circles). Similar results can be observed by using amyl acetate instead of acetone indicating that for larger amounts of the sample the solvent exchange process with liquid CO2 takes more time. These results show that the MOF/liquid_CO2 ratio as well as resting time in liquid CO2 (or corresponding CO2 exchange frequency) have a crucial impact on the textural properties of DUT-32.
It could be shown that by systematically extending the resting time in liquid CO2 (and exchange frequency), the porosity of DUT-32 can be significantly improved, finally reaching the theoretical limit (Fig. 2). The nitrogen uptake at 77 K increases steadily from 935 cm3 g−1 (one day in liquid CO2) up to a maximum of 2040 cm3 g−1 (seven days in liquid CO2). The maximum value corresponds to a specific pore volume of 3.16 cm3 g−1, which is remarkable. A further increase of exchange time to ten days showed only a slight porosity improvement with an adsorbed volume of nitrogen of 2080 cm3 g−1.
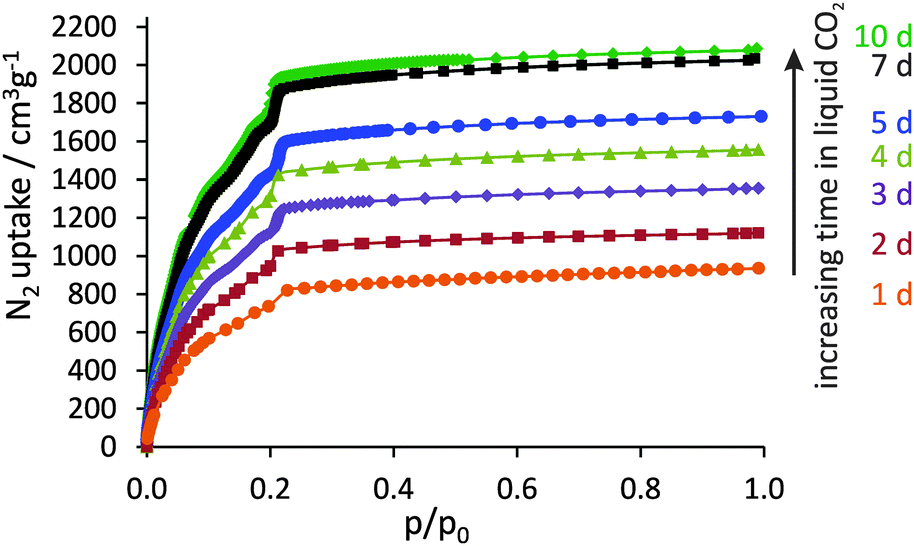 | ||
| Fig. 2 N2 physisorption isotherms at 77 K of DUT-32 after supercritical drying from amyl acetate as solvent with different resting times in liquid CO2 during the drying process. | ||
Applying consistency criteria suggested by Rouquerol and Llewellyn10 to the N2 physisorption isotherm for a sample with a resting time of seven days in liquid CO2 (further denoted as DUT-32_7d) results in an apparent BET surface area as high as 6411 m2 g−1 (see ESI†). This value is close to the geometrical surface area of DUT-32 calculated using the Poreblazer V1.2 program11 (5981 m2 g−1). Additionally, the nitrogen adsorption isotherm was simulated using multipurpose simulation code MUSIC.12 The simulated isotherm fits well with the experimental one (Fig. S13, ESI†). By applying the BET theory to the simulated isotherm in the same relative pressure region as for the experimental one (p/p0 0.09–0.13), the surface area of 6255 m2 g−1 was calculated.
DUT-32 also has excellent gas storage properties. For H2 the excess maximum capacity is achieved at 53 bar and reaches 85 mg g−1 (7.8 wt%; 22.9 mg cm−3) (Fig. 3) which is only surpassed by MOF-210 (86 mg g−1)7 and NU-100 (99.5 mg g−1).13 Considering the enormous pore volume, the total capacity at 80 bar corresponds to 166 mg g−1 (14.21 wt%; 44.9 mg cm−3) which is among the highest gravimetric values reported for porous materials.
 | ||
| Fig. 3 H2 (a, 77 K) and CH4 (b, 298 K) physisorption isotherms of DUT-32_7d; excess (●), total (◆); filled and empty symbols correspond to the adsorption and desorption, respectively. | ||
Recently Yildirim et al.1c claimed that a hypothetical MOF with optimal methane storage properties should have a pore volume of 3.2 cm3 g−1, a density of 0.27 g cm−3 and a surface area of 7500 m2 g−1 to be able to adsorb 0.5 g methane per gram of MOF (195 cc (STP) cc−1) at 65 bar. Interestingly, DUT-32 has the porosity characteristics which are very close to those of the desired hypothetical MOF: pore volume of 3.16 cm3 g−1 and density of 0.27 g cm−3. DUT-32's maximum excess methane capacity at 298 K is reached with 0.24 g g−1 at 105 bar (corresponding to 65 mg ml−1). Due to the large pore volume, DUT-32 achieves a total desired CH4 storage capacity of 0.55 g g−1 (149 g L−1, 210 cm3 (STP) cm−3) at 130 bar.
Summarizing, we have presented a new metal–organic framework (DUT-32) with a rare umt topology that can be easily scaled up due to the simple linker synthesis using cheap starting materials. Careful adjustment of the activation conditions revealed an ultrahigh porosity with a nitrogen uptake of 2080 cm3 g−1 (STP) corresponding to a total pore volume of 3.16 cm3 g−1 and an apparent BET area of 6411 m2 g−1, adding this compound to the top ten materials with the highest BET surface area.
The authors are grateful for the financial support from the DFG (SPP 1362), and the HZB. Special thanks goes to Dr G. Auffermann for carrying out the elemental analyses, and Dr I. Baburin for simulations.
Notes and references
- (a) H. Frost, T. Düren and R. Q. Snurr, J. Phys. Chem. B, 2006, 110, 9565 CrossRef CAS PubMed; (b) H. Frost and R. Q. Snurr, J. Phys. Chem. C, 2007, 111, 18794 CrossRef CAS; (c) Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 11887 CrossRef CAS PubMed; (d) J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32 RSC.
- (a) X. Song, Y. Zou, X. Liu, M. Oh and M. S. Lah, New J. Chem., 2010, 34, 2396 RSC; (b) R. Ma, C. Chen, B. Sun, X. Zhao and N. Zhang, Inorg. Chem. Commun., 2011, 14, 1532 CrossRef CAS PubMed; (c) S. Banerjee, N. N. Adarsh and P. Dastidar, Soft Matter, 2012, 8, 7623 RSC; (d) Y. Zhang, Q. Wang, Y.-J. Xiao, J. Han and X.-L. Zhao, Polyhedron, 2012, 33, 127 CrossRef PubMed; (e) L. Rajput, D. Kim and M. S. Lah, CrystEngComm, 2013, 15, 259 RSC; (f) Y.-Q. Chen, S.-J. Liu, Y.-W. Li, G.-R. Li, K.-H. He, Z. Chang and X.-H. Bu, CrystEngComm, 2013, 15, 1613 RSC.
- K. Koh, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2010, 132, 15005 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed.
- H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef CAS PubMed.
- K. Koh, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2009, 131, 4184 CrossRef CAS PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 CrossRef CAS PubMed.
- J. E. Mondloch, O. Karagiaridi, O. K. Farha and J. T. Hupp, CrystEngComm, 2013, 15, 9258–9264 RSC.
- A. W. Francis, J. Phys. Chem., 1954, 58, 1099 CrossRef CAS.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2007, 160, 49 CrossRef CAS.
- L. Sarkisov and A. Harrison, Mol. Simul., 2011, 37(14), 1248 CrossRef CAS; A. Gupta, S. Chempath, M. J. Sanborn, L. A. Clark and R. Q. Snurr, Mol. Simul., 2003, 29, 29 CrossRef.
- A. Gupta, S. Chempath, M. J. Sanborn, L. A. Clark and R. Q. Snurr, Mol. Simul., 2003, 29, 29 CrossRef CAS.
- O. K. Farha, A. Özgür Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details, single crystal X-ray structure analysis details, PXRD patterns, thermogravimetric analysis and NMR data. CCDC 968887 and 968888. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc00113c |
‡ Crystallographic data: C252.5H491.3N68O43.7Zn4, M = 5441.09 g mol−1, hexagonal, P63/m (Nr. 176), a = 50.710(7) Å, c = 62.920(13) Å, V = 140 122(40) Å3, Z = 18, ρcalc. = 1.161 g cm−3, λ = 0.88561 Å, T = 293 K, 2θmax = 28.2°, reflections collected/unique 118![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 656/60048, Rint = 0.0497, R1 = 0.0688, wR2 = 0.1815, S = 1.010, largest diff. peak 0.229 e Å−3 and hole −0.196 e Å−3. CCDC 968887. 656/60048, Rint = 0.0497, R1 = 0.0688, wR2 = 0.1815, S = 1.010, largest diff. peak 0.229 e Å−3 and hole −0.196 e Å−3. CCDC 968887. |
| This journal is © The Royal Society of Chemistry 2014 |