High-turnover visible-light photoreduction of CO2 by a Re(I) complex stabilized on dye-sensitized TiO2†
Eun-Gyeong
Ha
a,
Jeong-Ah
Chang
a,
Sung-Min
Byun
a,
Chyongjin
Pac
*b,
Dong-Myung
Jang
a,
Jeunghee
Park
*a and
Sang Ook
Kang
*a
aDepartment of Advanced Materials Chemistry, Korea University, 2511 Sejong-ro, Sejong-city 339-700, Korea. E-mail: parkjh@korea.ac.kr; sangok@korea.ac.kr
bYulchon Research Center, Korea University, Sejong-ro 2511, Sejong-city 339-700, Korea. E-mail: jjpac@korea.ac.kr
First published on 11th March 2014
Abstract
Hybrid systems prepared by fixing a Re(I) complex and a dye on three types of TiO2 nanoparticles in two different ways commonly revealed persistent photocatalysis of the CO2 reduction to CO with no levelling-off tendency under visible-light irradiation in DMF, giving a turnover number of ≥435.
The visible-light photoreduction of CO2 has been receiving considerable interest not only from environmental and long-term energy-security viewpoints,1 but also as a crucial scientific issue in “artificial photosynthesis”.2,3 An essential requirement for CO2 reduction in artificial photosynthesis is to couple the visible-light-driven flow of electrons with multiple-electron chemical processes that can lead to the formation C–H and C–C bonds and can cleave C–O bonds. Such processes require suitable catalysts, among which transition-metal complexes have been regarded as a potential candidate in artificial photosynthesis4 as well as in electrochemistry,5 due to the easy tuning of redox potentials, the trapping of CO2via its coordination to the metal centre, and the valence jump of the metal oxidation state in response to the multiple-electron reduction processes.
Among a variety of metal complexes investigated,2–5 the (bpy)ReI(CO)3Cl (bpy = 2,2′-bipyridine) complex reported by Lehn and his coworkers in 1983 is of particular interest because of the highly selective photoreduction of CO2 to CO in a relatively high quantum yield.6 Since then, related Re(I) complexes have been extensively applied to photochemical7 and electrochemical8 reductions of CO2. However, a serious drawback of the Re(I) complexes in the homogeneous-solution photocatalysis is their short durability, as revealed by leveling-off tendencies in the CO formation at a relatively early stage7,9 due to degradation of the Re(I) complexes.10 However, the degradation might not arise from the inherent properties of the Re(I) complexes, since high turnover numbers were reported for particular systems using sensitizer-bridged Re(I) supra-molecules11 or a sensitizer of cyclic Re(I) trimer and a Re(I) catalyst.12 In homogeneous solutions, various intermediates9,13 involving long-lived reactive species are unintentionally distributed to undergo intermolecular interactions associated with the degradation of the starting complex. It can therefore be expected that the fixing of a Re(I) complex on dye-sensitized semiconductors might allow each Re(I) molecule to work as an intrinsic catalyst under visible-light sensitization by the dye. An additional benefit of utilizing such semiconductor-based hybrid systems would derive from the potential capability of semiconductor materials in multiple-electron deposition and transfer to a catalyst site.3,14 This communication reports that hybrid systems prepared by immobilizing the Re(I) complex ReC on dye-loaded TiO2 nanoparticles reveal persistent behaviour in the photoreduction of CO2 to CO under visible-light irradiation in the presence of an electron donor (SD) (Fig. 1), and some interesting findings from this work are presented.
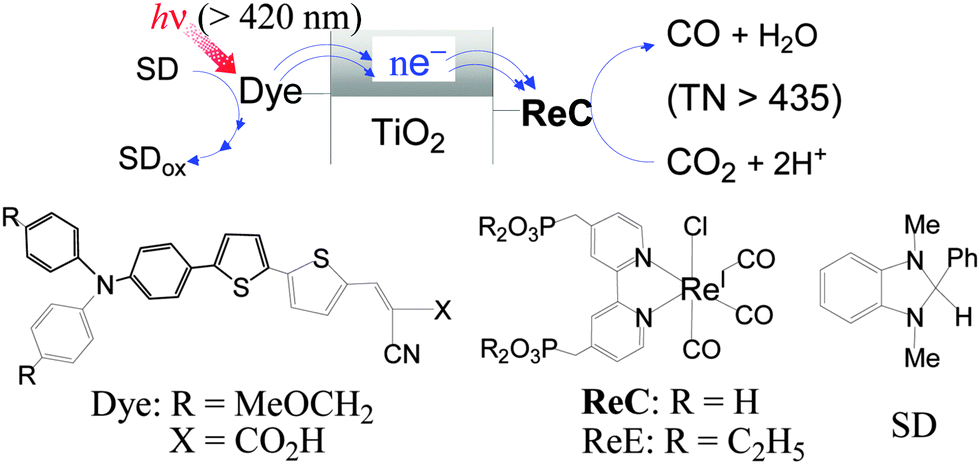 | ||
| Fig. 1 Conceptual representation for the reaction system (top) and the compounds used in this work (bottom). | ||
The organic dye (Dye), Re(I) catalysts (ReC and ReE) and electron donor (SD) shown in Fig. 1 were prepared according to known methods (see ESI†). The TiO2 materials used include synthetic nanosheets with exposed [001] facets (S-TiO2) and two commercially available nanosize powders, Hombikat UV-100 (H-TiO2) and Degussa P-25 (D-TiO2), onto which ReC and Dye were covalently fixed through the phosphonic or carboxylic acid anchoring group. The hybrid materials are denoted as ReC/TiO2/Dye when prepared by the initial loading of ReC followed by fixing of the Dye and as Dye/TiO2/ReC, which was obtained by the reverse sequence of loading. The successful anchoring of ReC and Dye on the TiO2 surface was confirmed by IR and diffuse-reflectance absorption spectroscopy (Fig. S1†). Suspensions of the hybrid materials in CO2-saturated N,N-dimethylformamide (DMF) containing SD (0.1 M) were irradiated at >420 nm using a Xenon lamp combined with a glass light filter.
As shown in Fig. 2(A) and Fig. S2† for the plots of TN(CO) (=molar ratio of CO formed/ReC used) versus irradiation time, CO was steadily formed with no levelling-off tendency; H2 evolution was only <5% that of CO. Table 1 lists TN(CO) after 10 h of irradiation. In the case of ReC/H-TiO2/Dye, the steady CO formation continued for 20 h with TN(CO) of 160 (Fig. S3†), and no substantial loss in the IR absorption bands of the CO ligands of ReC was observed (Fig. 2(B)). Formic acid and oxalic acid as the other possible reduction products were not detected upon HPLC analysis of the liquid phase.
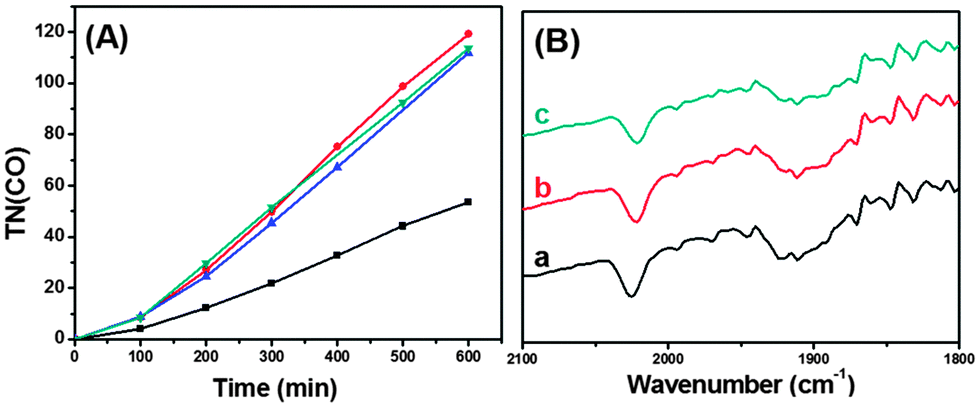 | ||
Fig. 2 (A) Plots of CO formation versus time in the absence (–■–) and presence of 1.5 M 2,2,2-trifluoroethanol ( ), 3% (v/v) H2O ( ), 3% (v/v) H2O ( ), and 10% (v/v) H2O ( ), and 10% (v/v) H2O ( ) for 10 mg ReC (0.1 μmol)/H-TiO2/Dye (1.5 μmol) in 3 mL DMF containing 0.1 M SD; irradiation at >420 nm. (B) IR spectra of ReC/H-TiO2/Dye in KBr discs (sample: KBr ≈ 1 ) for 10 mg ReC (0.1 μmol)/H-TiO2/Dye (1.5 μmol) in 3 mL DMF containing 0.1 M SD; irradiation at >420 nm. (B) IR spectra of ReC/H-TiO2/Dye in KBr discs (sample: KBr ≈ 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100) before (a) and after irradiation for 100 min (b) and for 20 h (c). :100) before (a) and after irradiation for 100 min (b) and for 20 h (c). | ||
For comparison, the photoreduction of CO2 in homogeneous DMF solution using a combination of ReE/triethanolamine (TEOA), ReE/SD or Ru(bpy)32+/ReE/SD was undertaken; the CO formation leveled off within 5 h with TN(CO) of <50 (Fig. S4†). These results clearly demonstrate that ReC has been remarkably stabilized by its fixation onto the Dye-loaded TiO2 nanoparticles. Presumably, two-electron transfers to the ReC site would be effectively mediated through TiO2 to complete the catalytic cycle without significant degradation of ReC. On the other hand, in homogeneous solutions, the second electron transfer to a key intermediate following the first one-electron reduction event should proceed under direct interactions with the second electron source(s), typically the one-electron reduced species of the Re(I) catalyst9 and oxidized donor radicals, so that unfavourable competitive reactions might occur even to a minor extent.
While the electron flow in the CO2 reduction should follow the scheme shown in Fig. 1, the relative energy levels of the components need to be referred to. The flat-band potential of a fused-particle TiO2 electrode in DMF was reported to lie at −2.04 V versus SCE (−2.42 V versus Fc+/0),15 which is 0.42 eV more negative than the oxidation potential of excited-singlet Dye (1Dye*).16 If this were the case, the electron injection from 1Dye* into the conduction band of TiO2 would hardly compete with the decay of 1Dye* (τ ≈ 1 ns).16 Furthermore, the electron transfer to ReC through TiO2 should be exergonic enough to compete with fast charge recombination.17 Therefore, the conduction-band minimum of our TiO2 materials in DMF might be located between the oxidation potential of 1Dye* (≈−2 V versus Fc+/0)17 and the reduction potential of ReC (−1.67 V). The oxidation potential of SD (−0.185 V) is much more negative than that of Dye (0.50 V) so that the Dye radical cation left after electron injection from 1Dye* into TiO2 might be efficiently reduced by SD.
Some interesting features from the present observations should be noted. (1) The photocatalytic efficiencies significantly depend on the TiO2 sources (Table 1), probably related, at least in part, to the different morphologies and crystal phases of the nanoparticles (Fig. S5 and S6†). S-TiO2 has an anatase nanosheet morphology (20 nm length × 5 nm thickness) with ≈90% [001] facets, whereas H-TiO2 (pure anatase) and D-TiO2 (75% anatase and 25% rutile) are spherical nanoparticles of 5 nm and 18 nm diameter, respectively. The agglomerates of the different nanoparticles should have different distributions of surface states/trap sites and grain boundaries associated with the catalytic properties. (2) In the cases of S- and D-TiO2, the initial loading of Dye resulted in considerably higher catalytic activity than the initial loading of ReC, whereas such a loading-sequence effect was not clear in the case of H-TiO2 (Table 1 and Fig. S1†). A possible assumption is that the surfaces of larger-size S- and D-TiO2 would have local distributions with different activities in the electron injection from 1Dye* and/or electron transfer to ReC, while the surface of smaller-size pure anatase H-TiO2 would be relatively homogeneous. (3) The reaction efficiencies were considerably enhanced on addition of 1.5 M 2,2,2-trifluoroethanol or 3–10% (v/v) H2O, particularly in the cases of the hybrids based on H-TiO2 and D-TiO2 (Table 1). This observation is reminiscent of the Brønsted acid effect on electrochemical CO2 reduction.18 Such an effect was minor, but appreciable, for the S-TiO2 hybrids. (4) The CO2 reduction was almost completely retarded by 1.2 M TEOA, unlike homogeneous-solution CO2 reduction which is more or less assisted by TEOA coexisting with a real electron donor.11,12
In order to confirm the catalytic persistency of the hybrids, we carried out repetitive irradiation experiments. As shown in Fig. 3 for Dye/H-TiO2/ReC, no leveling-off tendency was observed in each cycle and the efficiency of CO formation increased with the increase of the cycle from a TN(CO) of 84 in the 1st run to a TN(CO) of 121 in the 4th run. The total TN(CO) reached 435. The other hybrids also revealed similar behaviour as well (Fig. S8†). This unique phenomenon appears to be in line with the appearance of induction periods in Fig. 2, Fig. S2–S4, S7, and S8.† While investigations are now being performed on the mechanistic origin, we tentatively assume that the electron transfer to ReC would progressively prevail over electron trapping as various electron-trapping sites distributed in TiO217,19 have been sequentially filled with trapped electrons.
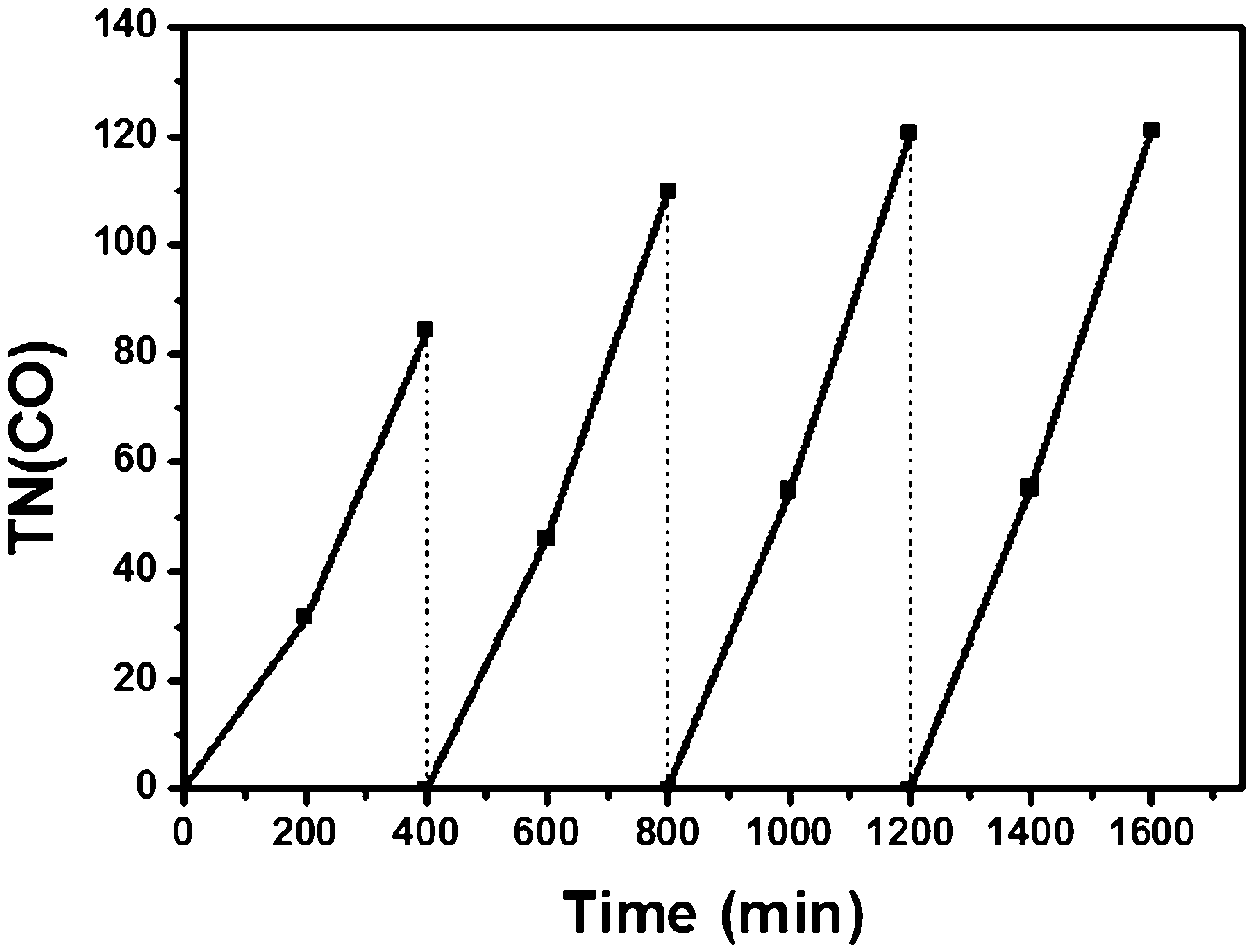 | ||
| Fig. 3 Formation of CO in a 4-cycle repetition of irradiation at >420 nm for 400 min after CO2 bubbling for 30 min in the dark; 10 mg Dye/S-TiO2/ReC with 0.1 μmol ReC and 1.5 μmol Dye in the presence of 0.1 M SD and 10% (v/v) H2O. | ||
The present investigation has demonstrated that the ReC catalyst immobilized on Dye-sensitized TiO2 particles works as a persistent catalyst for the reduction of CO2 to CO with high TN(CO) under visible-light irradiation. It is implied that the convenient methodology reported here might provide a possible way for manifesting the “inherent” catalytic ability of particular transition-metal complexes that would be masked in homogeneous-solution catalysis. A further attempt is currently being made to find stable hybrid systems that can efficiently work in water, directed at coupling the CO2 reduction hybrid with a water–oxidation system.
This research was supported by a Korea University Grant.
Notes and references
- S. Styring, Faraday Discuss., 2012, 155, 357 RSC; A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. Dubois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. Kenis, C. A. Kerfeld, R. H. Morris, C. H. Peden, A. R. Portis, S. R. Ragasdale, T. B. Rauchfues, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621 CrossRef CAS PubMed.
- T. Yui, Y. Tamaki, K. Sekigawa and O. Ishitani, Top. Curr. Chem., 2011, 303, 151 CrossRef CAS PubMed.
- L. Alibabai, H. Luo, R. L. House, P. G. Hoeltz, R. Lopez and T. J. Meyer, J. Mater. Chem. A, 2013, 1, 4133 Search PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983 CrossRef CAS PubMed; J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036 RSC.
- K. Tanaka and D. Ooyama, Coord. Chem. Rev., 2002, 226, 211 CrossRef CAS; J.-M. Savéant, Chem. Rev., 2008, 108, 2348 CrossRef PubMed; E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89 RSC; C. Constentin, M. Robert and J.-M. Savéant, Chem. Soc. Rev., 2013, 42, 2423 RSC.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536 RSC; J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990 CrossRef CAS.
- H. Takeda, K. Koike, T. Morimoto, H. Inumaru and O. Ishitani, Adv. Inorg. Chem., 2011, 63, 137 CrossRef CAS; H. M. Sung-Suh, D. S. Kim, C. W. Kim, C. W. Lee and S.-E. Park, Appl. Organomet. Chem., 2000, 14, 826 CrossRef; P. Kurz, B. Probst, B. Spingler and R. Alberto, Eur. J. Inorg. Chem., 2006, 2966 CrossRef; C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445 CrossRef PubMed; C. Liu, K. D. Dubois, M. E. Lois, A. S. Vorushilov and G. Li, ACS Catal., 2013, 3, 655 CrossRef; G. A. Andrade, A. J. Pistner, G. P. A. Yap, G. A. Lutterman and J. Rosenthal, ACS Catal., 2013, 3, 1685 CrossRef PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328 RSC; J. R. O'toole, L. D. Margelum, T. D. Westmoreland, W. J. Vining, R. W. Murray and T. J. Meyer, J. Chem. Soc., Chem. Commun., 1985, 1416 RSC; B. K. Kumar, J. M. Smieja, A. F. Sasayama and C. P. Kubiak, Chem. Commun., 2012, 48, 272 RSC.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023 CrossRef CAS PubMed.
- C. Kutal, M. A. Weber, G. Ferraudi and G. Geiger, Organometallics, 1985, 4, 2161 CrossRef CAS; C. Kutal, A. J. Corbin and G. Ferraudi, Organometallics, 1987, 6, 553 CrossRef; O. Ishitani, I. Namura, S. Yanagida and C. Pac, J. Chem. Soc., Chem. Commun., 1987, 1153 RSC.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326 CrossRef CAS PubMed; Z.-Y. Bian, K. Sumi, M. Furue, S. Sato, K. Koike and O. Ishitani, Dalton Trans., 2009, 983 RSC; Y. Tamaki, K. Watanabe, K. Koike, H. Inoue and O. Ishitani, Faraday Discuss., 2012, 155, 115 RSC; Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22 CrossRef.
- T. Morimoto, C. Nishiura, M. Tanaka, J. Rohacova, Y. Nakagawa, Y. Funada, K. Koike, Y. Yamamoto, S. Shishido, T. Kojima, T. Saeki, T. Ozeki and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 13266 CrossRef CAS PubMed.
- B. P. Sullivan and T. J. Meyer, J. Chem. Soc., Chem. Commun., 1984, 1244 RSC; B. P. Sullivan, C. M. Bolinger, D. Conrad, W. J. Vining and T. J. Meyer, J. Chem. Soc., Chem. Commun., 1985, 1414 RSC; D. H. Gibson, X. Yin, H. He and M. S. Mashuta, Organometallics, 2003, 22, 337 CrossRef CAS; Y. Hayashi, S. Kita, B. S. Brunschwig and E. Fujita, J. Am. Chem. Soc., 2003, 125, 11976 CrossRef PubMed; K. D. Dubois, A. Petushkov, E. G. Cardona, S. C. Larsen and G. Li, J. Phys. Chem. Lett., 2012, 3, 486 CrossRef PubMed.
- T. W. Woolerton, S. Sheard, F. Reisner, E. Pierce, S. W. Ragasdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 132, 2123 CrossRef CAS PubMed; Y. S. Chaudhary, T. W. Woolerton, C. S. Allen, T. H. Warner, E. Pierce, S. W. Ragasdale and F. A. Armstrong, Chem. Commun., 2012, 48, 58 RSC.
- G. Redmond and D. F. Fitzmaurice, J. Phys. Chem., 1993, 97, 1426 CrossRef CAS.
- S.-H. Lee, Y. Park, K. R. Wee, H. J. Son, D. W. Cho, C. Pac, W. Choi and S. O. Kang, Org. Lett., 2010, 12, 460 CrossRef CAS PubMed.
- W.-S. Han, K.-R. Wee, H.-Y. Kim, C. Pac, Y. Nabetani, D. Yamamoto, T. Shimada, H. Inoue, H. Choi, K. Cho and S. O. Kang, Chem. – Eur. J., 2012, 18, 16368 Search PubMed.
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283 CrossRef CAS PubMed.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr., Chem. Rev., 1995, 95, 735 CrossRef CAS; J. R. Durrant, S. A. Haque and J. R. Plomares, Coord. Chem. Rev., 2004, 248, 1247 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, IR spectra and DRS of ReC/H-TiO2/Dye, plots of TN(CO) versus time for the hybrid systems and SEM images of the TiO2 sources. See DOI: 10.1039/c3cc49744e |
| This journal is © The Royal Society of Chemistry 2014 |