 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective iron-catalyzed alkyne hydrogenation in ionic liquids†
Tim N.
Gieshoff
a,
Alice
Welther
a,
Michael T.
Kessler
b,
Martin H. G.
Prechtl
*b and
Axel
Jacobi von Wangelin
*a
aInstitute of Organic Chemistry, University of Regensburg, Germany. E-mail: axel.jacobi@ur.de; Fax: +49 (0)941 943 4617
bDept. of Chemistry, University of Cologne, 50939 Cologne, Germany
First published on 17th January 2014
Abstract
Iron(0) nanoparticles in ionic liquids (ILs) have been shown to catalyse the semi-hydrogenation of alkynes. In the presence of a nitrile-functionalised IL or acetonitrile, stereoselective formation of (Z)-alkenes was observed. The biphasic solvent system allowed facile separation and re-use of the catalyst.
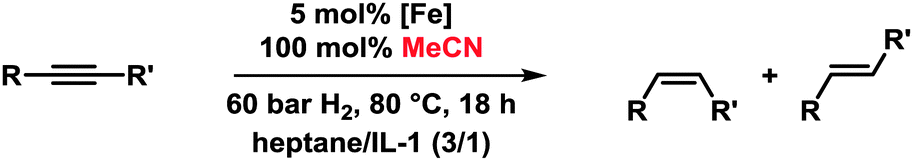
Iron-catalysed hydrogenations are among the largest technical processes (Haber–Bosch, gas-to-liquid)1 but are under-utilised on the smaller scales of fine chemical, agrochemical, and pharmaceutical manufacture and within academic synthesis programs. However, the current economic and environmental constraints have prompted reconsiderations of iron-catalysed procedures.2 Hydrogenations of alkenes and alkynes with well-defined ligand-stabilised iron catalysts or heterogeneous species have been recently reported.3 Hydrogenations of alkynes in general bear the dual challenge of product- and stereo-selectivity. Lindlar-type catalysts exhibit especially high versatility and are the benchmark for semi-hydrogenations of (Z)-alkenes.4 This combination of an expensive noble metal catalyst (Pd/CaCO3) and toxic additives (Pb(OAc)2, quinoline) is clearly derogatory to the development of sustainable chemical processes, so an inexpensive nontoxic iron-catalysed alternative is highly desirable.5 However, the search for new catalysts for technical applications is incomplete without the implementation of efficient catalyst separation and recycling technologies.6 Despite the higher selectivity of homogeneous catalysts, most technical processes use heterogeneous catalysts because of their ease of separation from the products. Within the scope of our iron catalysis program, we thus aimed at merging the benefits of a separable heterogeneous catalyst with that of a highly dispersive ligand-modified catalyst. We envisioned to capitalise on the hybrid concept of nanoparticular iron catalysts7 in the presence of a ligand which controls the catalyst selectivity through coordination and an ionic liquid to allow catalyst separation. The potential of such modular systems was evaluated in stereoselective semi-hydrogenations of alkynes (Scheme 1).
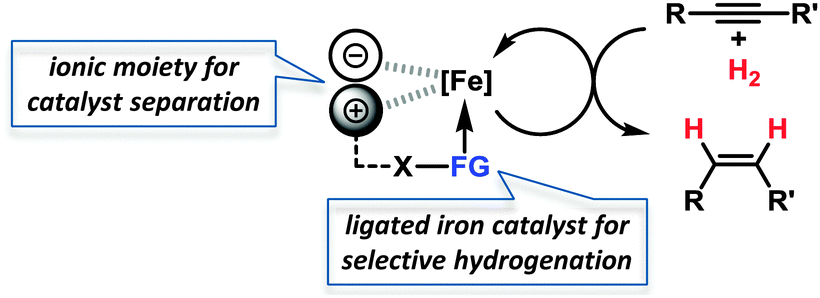 | ||
| Scheme 1 Modular ion pair/ligand/iron catalysts for hydrogenations. | ||
Ionic liquids (ILs) based on azolium salts seemed to be perfectly suited for this task due to their ability to dissolve, stabilise, and modulate metal nanoparticles.8 Their physicochemical properties are widely adjustable by variation of substituents, the heterocycle, and counterions. Most importantly, azolium-ILs are immiscible with non-polar solvents and have a negligible vapour pressure. IL-stabilised precious metal nanoparticle catalysts were successfully applied to hydrogenations whereas the generation and utilization of IL-embedded iron catalysts are as yet under-utilised.9
We set out to study the catalytic activity of heterogeneous iron species in low oxidation states generated by reduction of FeCl3 with ethylmagnesium chloride (Table 1).3c,e Hydrogenation of diphenylacetylene (1) in the absence of a suitable ligand showed low selectivity toward stilbene (2). The use of imidazolium ILs generally required elevated pressure and temperature. Geared by literature precedents with Pd, Ru, and Au nanoparticles, we employed nitrile-functionalised ILs as catalyst modifiers.9,10 Notably, the presence of a nitrile function, the suppression of N-heterocyclic carbene formation,11 and the absence of THF were crucial to a selective cis-hydrogenation in IL-3.5 The replacement of THF with heptane also resulted in high phase-partitioning at room temperature and allowed facile separation of the catalyst. The higher reaction temperature is believed to enhance dispersion of the viscous IL with the non-polar phase.
|
|
||||
|---|---|---|---|---|
| Entry | Conditionsb | Solvent(s) | 1/2/3c [%] | Z/E (2)c |
| a [Fe]: 5 mol% FeCl3, THF, 20 mol% EtMgCl, THF, r.t., 30 min. b For experimental details, see ESI. c Determined by 1H-NMR and GC-FID. d Removal of THF prior to reaction. | ||||
| 1 | 4 bar H2, 45 °C, 8 h | THF | 0/2/93 | n.d. |
| 2 | 1 bar H2, 18 °C, 8 h | THF | 1/73/21 | 3/1 |
| 3 | 50 bar H2, 50 °C, 16 h | THF–IL-1 | 2/3/93 | n.d. |
| 4 | 4 bar H2, 18 °C, 16 h | THF–IL-2 | 94/2/2 | n.d. |
| 5 | 50 bar H2, 50 °C, 16 h | THF–IL-2 | 82/15/2 | 6/1 |
| 6 | 50 bar H2, 50 °C, 60 h | THF–IL-3 | 23/75/1 | 7/1 |
| 7 | 60 bar H2, 80 °C, 44 h | hept–IL-3 | 1/94/3 | 19/1 |
| 8 | 30 bar H2, 80 °C, 20 h | hept–IL-1d | 0/1/98 | n.d. |
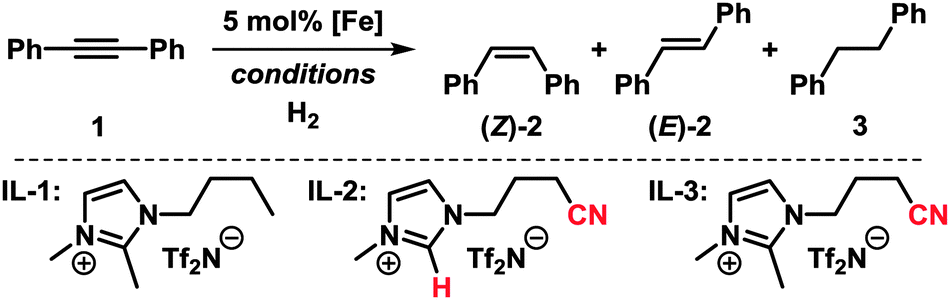
Application of the optimised conditions in a biphasic heptane–IL-3 mixture to other phenylacetylenes resulted in moderate to good yields of the corresponding alkenes and generally high stereoselectivities toward the (Z)-isomers (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | Alkyne | R | Yield [%] | Z/E |
| a For experimental details, see ESI. | ||||
| 1 |
|
H | 94 | 95/5 |
| 2 | 4-tBu | 86 | 95/5 | |
| 3 | 4-OMe | 92 | 93/7 | |
| 4 | 4-F | 75 | 100/0 | |
| 5 | 3-OMe | 52 | 96/4 | |
| 6 |
|
68 | 89/11 | |
| 7 |
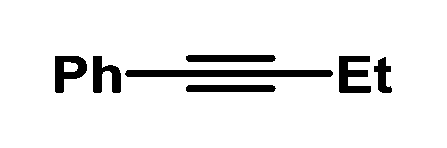
|
80 | 88/12 | |
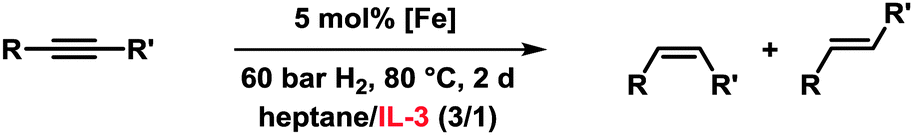
Our observation that ILs can effectively stabilise nanoparticles is in full accord with literature reports.9,10 Droplets of Fe-NPs in IL-1 and IL-3 were measured by transmission electron microscopy (TEM), respectively (Fig. 1). Both species are approximately 4–5 nm in diameter and slowly grow during the catalytic reactions (to ∼8–20 nm after 24 h under hydrogenation reaction conditions).
 | ||
| Fig. 1 Particle sizes and TEM image of Fe@IL catalysts. | ||
From a mechanistic point of view, the presence of two electrophilic functions (imidazolium cations and nitrile groups) and a strong base/nucleophile (EtMgCl) poses the question of the actual nature of the ligand moiety that is present under the reaction conditions. We treated IL-1 with 1 equiv. EtMgCl at room temperature for 30 min followed by quenching with deuterium oxide (D2O) which afforded a mixture of ring-deuterated (35%) and 2-deuteromethyl products (10%).12 IL-3 did not undergo deuteration under identical conditions (2H-NMR, MS). Instead, a dimer was formed which was detected by GC-MS and ESI-MS and tentatively assigned as shown in Scheme 2.13 If a nucleophile-assisted manipulation of the nitrile is relevant to the control of selectivity, simple alkylnitriles should exert a similar effect. We therefore examined whether the intramolecular bifunctional motif of IL-3 could also be expressed by a much simpler intermolecular combination of ionic liquid and nitrile functions. Furthermore, the potential occurrence of acetyl or imine moieties as degradation products of the nitrile prompted us to also investigate the activity of ternary catalyst systems comprising a pre-formed reduced iron species (5 mol%), the non-functionalised ionic liquid IL-1, and various carbonyl derivatives under the optimised conditions (Scheme 3 and Table 3). Without additives, the hydrogenation of 1 in heptane–IL-1 afforded dibenzyl (3, >95% yield, Table 3, entry 1).
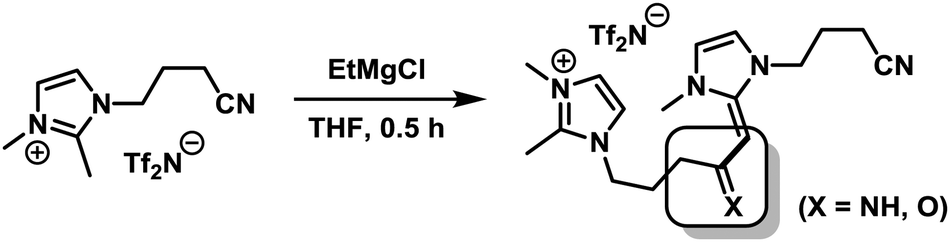 | ||
| Scheme 2 Formation of a dimer of IL-3 by treatment with EtMgCl. | ||
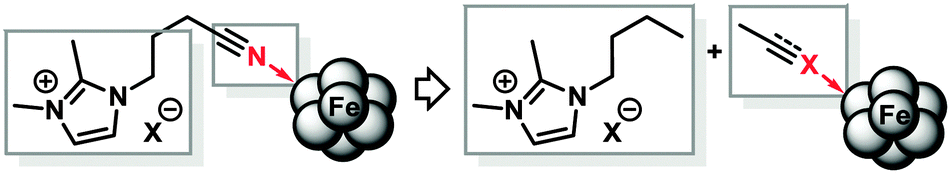 | ||
| Scheme 3 Intramolecular vs. intermolecular mode of bifunctionality. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Additive | Conversion [%] | 2 [%] | Z/E (2) | |
| a For experimental details, see ESI. b 100 mol% additive. c 20 bar H2. | |||||
| 1 | — | 100 | 3 | 67/33 | |
| 2 |
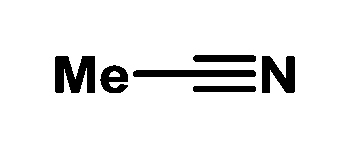
|
96 | 93 | 96/4 | |
| 3b,c | 99 | 85 | 96/4 | ||
| 4b | MeCN + PhCO2Me | 99 | 95 | 97/3 | |
| 5b | MeCN + PhCl | 100 | 95 | 94/6 | |
| 6b | MeCN + Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
100 | 95 | 96/4 | |
| 7b | MeCN + PhI | 43 | 38 | 95/5 | |
| 8b | MeCN + PhNO2 | 5 | 0 | — | |
| 9b |
|
R, R′ = Me | 25 | 22 | 91/9 |
| 10b | R, R′ = Ph | 100 | 84 | 94/6 | |
| 11 | R = Me, R′ = OEt | 92 | 86 | 95/5 | |
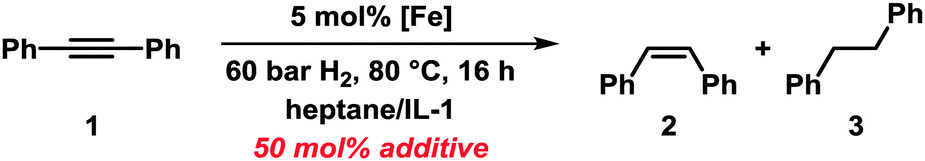
To our delight, identical productivity and stereoselectivity to those with the bifunctional IL-3 were observed when adding acetonitrile (entry 2), even at 20 bar H2 (entry 3). The loading of acetonitrile could be varied from 50 to 200 mol% without any change in selectivity. Further addition of 100 mol% methyl benzoate, chlorobenzene14 or 1,1-diphenylethylene, respectively, resulted in no change in activity (entries 4–6). Iodobenzene slowed down conversion while nitrobenzene acted as an inhibitor. Benzophenone and ethyl acetate showed only slightly lower activity and selectivity as MeCN (entries 10 and 11). Nanoparticles prepared from EtMgCl and EtMgBr afforded identical catalytic results.
The employment of a ternary Fe/IL-1/additive catalyst constitutes a significant simplification of the procedure and allows shorter reaction times than with the bifunctional IL-3 (16 h vs. 2 d). Table 4 shows selected examples of hydrogenations of various alkynes in the presence of 5 mol% iron catalyst and 100 mol% acetonitrile in the biphasic solvent mixture IL-1–n-heptane. Generally, higher yields and stereoselectivities were obtained compared with the reactions in IL-3 (Table 4). Free NH2 groups, esters, and alkenes were tolerated. Bulky (TMS) groups and carboxylates led to lower conversions. 1-Alkynes gave mixtures of alkenes and alkanes. Hydrogenations proceeded also in mono-phasic THF–MeCN or toluene–MeCN (40 h) mixtures with similar selectivity, but the catalyst phase could not be separated. The catalyst species was found to rapidly age in the absence of IL-1 and lose activity after 48 h. On the other hand, effective catalyst separations and multiple re-uses without loss of catalytic activity were realised with the ternary catalyst Fe/IL-1/MeCN in hydrogenations of 1 under standard conditions (Fig. 2). ICP-OES analysis of the product phase showed <0.03% leaching of iron (= 0.0015 mol%). Replacement of IL-1 with tetraalkyl-ammonium bromides (n-butyl, n-decyl) cleanly gave the alkanes, and product extraction with heptane failed. The heterogeneity of the catalyst species in IL-1/MeCN/heptane was further documented by the absence of inhibition upon addition of dibenzo[a,e]cyclooctatetraene (dct).15
|
|
||||
|---|---|---|---|---|
| Entry | Alkyne | R | Yieldb [%] | Z/E |
| a See ESI. b Conversion in parentheses if not >90%. | ||||
| 1 |
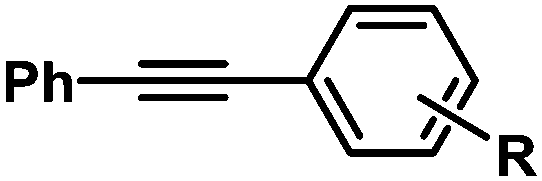
|
H | 97 | 96/4 |
| 2 | 4-tBu | 94 | 97/3 | |
| 3 | 4-OMe | 98 | 100/0 | |
| 4 | 4-NH2 | 76 | 100/0 | |
| 5 | 4-Br | 84 | 100/0 | |
| 6 | 4-Cl | 89 | 100/0 | |
| 7 | 4-F | 83 | 99/1 | |
| 8 | 4-CO2Me | 53 (74) | 100/0 | |
| 9 | 2-Cl | 79 | 100/0 | |
| 10 | 2-F | 82 | 100/0 | |
| 11 |
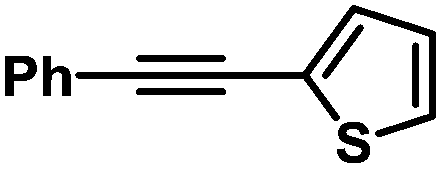
|
76 | 99/1 | |
| 12 |
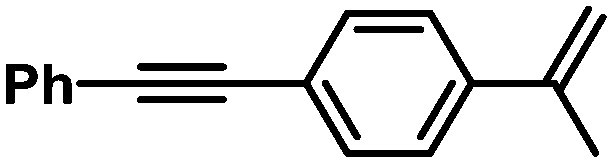
|
90 | 100/0 | |
| 13 |
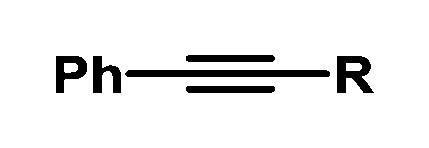
|
Et | 79 | 95/5 |
| 14 | TMS | 13 (19) | 96/4 | |
| 15 | CO2Me | 19 (40) | 92/8 | |
| 16 |
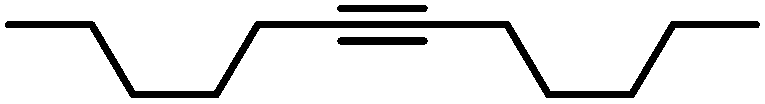
|
90 | 100/0 | |
| 17 |
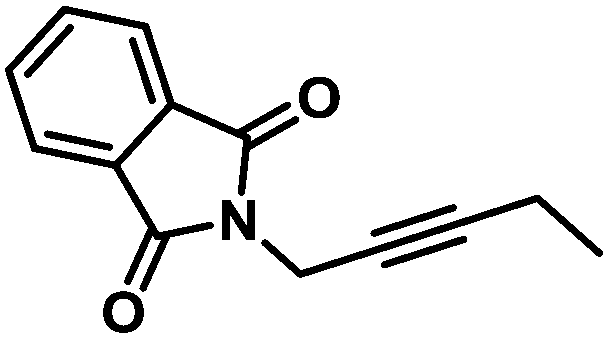
|
38 (70) | 100/0 | |
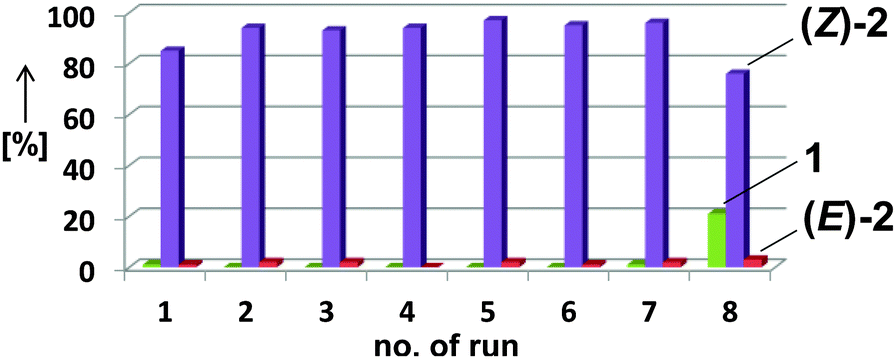 | ||
| Fig. 2 Consecutive hydrogenations of 1 with the identical catalyst phase after liquid–liquid decantation. | ||
In summary, we have developed a simple ternary iron catalyst that enables the (Z)-selective semi-hydrogenation of alkynes in a biphasic solvent mixture and the separation/reuse of the catalyst. The nanoparticle catalysts (∼5 nm) formed upon reduction of FeCl3 with EtMgCl. Acetonitrile effects stereocontrol; 1-butyl-2,3-dimethyl-1,3-imidazolium triflimide (IL-1) allows catalyst separation from the products and prevents particle aggregation.
This work was financed by the Ministry of Innovation, Science, Research (NRW-returnee fellowship to M.H.G.P.), the Evonik Foundation (fellowships to M.T.K., T.N.G.), the Robert-Lösch-Foundation, and the DFG. We thank Dr L. Greiner and S. Mariappan (Dechema) for performing TEM analyses.
Notes and references
- (a) G. Ertl, Catal. Rev. Sci. Eng., 1980, 21, 201–223 CrossRef CAS; (b) G. P. van der Laan and A. A. C. M. Beenackers, Catal. Rev. Sci. Eng., 1999, 41, 255–318 CrossRef CAS PubMed.
- (a) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed; (b) W. M. Czaplik, M. Mayer, J. Cvengros and A. Jacobi von Wangelin, ChemSusChem, 2009, 2, 396–407 CrossRef CAS PubMed.
- (a) S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794–13807 CrossRef CAS PubMed; (b) R. J. Trovitch, E. Lobkovsky, E. Bill and P. J. Chirik, Organometallics, 2008, 27, 1470–1478 CrossRef CAS; (c) P. H. Phua, L. Lefort, J. A. F. Boogers, M. Tristany and J. G. de Vries, Chem. Commun., 2009, 3747–3749 RSC; (d) M. Stein, J. Wieland, P. Steurer, F. Tölle, R. Mülhaupt and B. Breit, Adv. Synth. Catal., 2011, 353, 523–527 CrossRef CAS; (e) A. Welther, M. Bauer, M. Mayer and A. Jacobi von Wangelin, ChemCatChem, 2012, 4, 1088–1093 CrossRef CAS; (f) V. Kelsen, B. Wendt, S. Werkmeister, K. Junge, M. Beller and B. Chaudret, Chem. Commun., 2013, 49, 3416–3418 RSC.
- H. Lindlar, Helv. Chim. Acta, 1952, 35, 446–450 CrossRef CAS.
- (a) S. Enthaler, M. Haberberger and E. Irran, Chem.–Asian J., 2011, 6, 1613–1623 CrossRef CAS PubMed; (b) L. Ilies, T. Yoshida and E. Nakamura, J. Am. Chem. Soc., 2012, 134, 16951–16954 CrossRef CAS PubMed; (c) C. Belger and B. Plietker, Chem. Commun., 2012, 48, 5419–5421 RSC.
- Catalyst Separation, Recovery and Recycling, ed. D. J. Cole-Hamilton and R. P. Tooze, Springer, Dordrecht, 2006 Search PubMed.
- (a) D. L. Huber, Small, 2005, 1, 485–501 CrossRef PubMed; (b) A. Welther and A. Jacobi von Wangelin, Curr. Org. Chem., 2013, 17, 326–335 CrossRef CAS; (c) R. Hudson, G. Hamasaka, T. Osako, Y. M. A. Yamada, C.-J. Li, Y. Uozumi and A. Moores, Green Chem., 2013, 15, 2141–2148 RSC.
- (a) T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed; (b) N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC; (c) C. Vollmer and C. Janiak, Coord. Chem. Rev., 2011, 255, 2039–2057 CrossRef CAS PubMed; (d) P. S. Campbell, M. H. G. Prechtl, C. C. Santini and P. H. Haumesser, Curr. Org. Chem., 2013, 17, 414–429 CrossRef CAS; (e) M. H. G. Prechtl, J. D. Scholten and J. Dupont, Molecules, 2010, 15, 3441–3461 CrossRef CAS PubMed.
- (a) C. Janiak, Z. Naturforsch., B: J. Chem. Sci., 2013, 68, 1056–1089 Search PubMed; (b) J. D. Scholten, B. C. Leal and J. Dupont, ACS Catal., 2012, 2, 184–200 CrossRef CAS; (c) K. L. Luska and A. Moores, ChemCatChem, 2012, 4, 1534–1546 CrossRef CAS; (d) M. H. G. Prechtl, M. Scariot, J. D. Scholten, G. Machado, S. R. Teixeira and J. Dupont, Inorg. Chem., 2008, 47, 8995–9001 CrossRef CAS PubMed; (e) R. Venkatesan, M. H. G. Prechtl, J. D. Scholten, R. P. Pezzi, G. Machado and J. Dupont, J. Mater. Chem., 2011, 21, 3030–3036 RSC.
- (a) W. Zhu and Z. Hou, Curr. Inorg. Chem., 2013, 2, 213–227 CrossRef; (b) V. I. Parvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef CAS PubMed; (c) M. H. G. Prechtl, J. D. Scholten and J. Dupont, J. Mol. Catal. A: Chem., 2009, 313, 74–78 CrossRef CAS PubMed; (d) H. Wender, P. Migowski, A. F. Feil, L. F. de Oliveira, M. H. G. Prechtl, R. Leal, G. Machado, S. R. Teixeira and J. Dupont, Phys. Chem. Chem. Phys., 2011, 13, 13552–13557 RSC.
- D. Bézier, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2013, 355, 19–33 CrossRef.
- R. Giernoth and D. Bankmann, Eur. J. Org. Chem., 2008, 2881–2886 CrossRef CAS.
- C. E. I. Knappke, J. M. Neudörfl and A. Jacobi von Wangelin, Org. Biomol. Chem., 2010, 8, 1695–1705 CAS.
- Fe-catalysed dehalogenation: W. M. Czaplik, S. Grupe, M. Mayer and A. Jacobi von Wangelin, Chem. Commun., 2010, 46, 6350–6352 RSC.
- D. R. Anton and R. H. Crabtree, Organometallics, 1983, 2, 855–859 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc49679a |
| This journal is © The Royal Society of Chemistry 2014 |