 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA semiconducting microporous framework of Cd6Ag4(SPh)16 clusters interlinked using rigid and conjugated bipyridines†
Chao
Xu
ab,
Niklas
Hedin
*b,
Hua-Tian
Shi
a and
Qian-Feng
Zhang
*a
aInstitution of Molecular Engineering and Applied Chemistry, Anhui University of Technology, Ma'anshan, Anhui 243002, P. R. China. E-mail: zhangqf@ahut.edu.cn; Fax: +86-555-2312041; Tel: +86-55-2311059
bDepartment of Materials and Environmental Chemistry, Berzelii Center EXSELENT on Porous Materials, Arrhenius Laboratory, Stockholm University, Stockholm, SE-106 91, Sweden. E-mail: niklas.hedin@mmk.su.se; Tel: +46-8-162417
First published on 12th February 2014
Abstract
Ternary supertetrahedral chalcogenolate clusters were interlinked with bipyridines into a microporous semiconducting framework with properties qualitatively different from those of the original clusters. Both the framework and the clusters were effective photocatalysts, and rapidly degraded the dye rhodamine B.
Supertetrahedral chalcogenide and chalcogenolate clusters (SCCs) have intriguing structures and size-dependent semiconducting and photocatalytic properties.1,2 Their tetrahedral structure and multiple coordination sites allow them to assemble into extended and porous semiconductors with properties that are different from those of the individual clusters and related bulk materials. SCCs can be organized into extended frameworks via corner sharing, terminal coordination with extra metal ions, or organic linkers.3–7 Porous zeolite-like chalcogenides have been synthesized from SCCs by corner-sharing S2− groups. They were shown to act as both photocatalysts and hosts for hydrogen generation from water.8 An open metal-ion-coordinated framework of {Sn[Zn4Sn4S17]}6− is a reported example of an ion-exchange material.9 SCCs interlinked by multifunctional organic linkers are a class of metal–organic frameworks (MOF).10,11 Feng et al. co-assembled imidazoles and In–(Cd)–S SCCs into microporous compounds with relatively high capacities to adsorb CO2.12 We present here a ternary chalcogenolate cluster of Cd6Ag4(SPh)16(DMF)4 (1) (DMF: N,N′-dimethylformamide) and its three-dimensional microporous assembly of {[Cd6Ag4(SPh)16](bpe)2} (2) interlinked with rigid bpe (trans-1,2-bis(4-pyridyl)ethylene).
The discrete Cd6Ag4(SPh)16(DMF)4 cluster 1 was synthesized by the reaction of Cd(SPh)2 and AgNO3 in a solution of DMF. The organic groups surrounding the clusters stabilized the structure and increased their solubility. Cluster 1 was soluble in DMF, slightly soluble in dimethyl sulfoxide (DMSO), and insoluble in other common solvents. Further reactions of cluster 1 most probably ligated its terminal Cd atoms with the nitrogen donors of the bpe linker in DMF to afford the microporous framework 2. Large crystals of both the cluster 1 and the microporous framework 2 crystallized upon the slow evaporation of DMF and were structurally determined by single-crystal X-ray diffraction. Cluster 1 showed a supertetrahedral (T3) structure with ten metal centers (six Cd, four Ag) coordinated with four terminal DMF, six μ2-SPh, and four μ3-SPh ligands. The μ3-SPh ligand was not previously observed in SCCs. Balancing the charge of the neutral cluster suggests the presence of six Cd(II) and four Ag(I). The composition was confirmed by energy-dispersive X-ray spectroscopy, which gave a Cd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ag:S atomic ratio of 6.4:4:16.2 (Fig. S1, ESI†). Cd(II) and Ag(I) are isoelectronic; therefore, their relative positions within the clusters cannot be determined by X-ray diffraction. Their positions were instead inferred by applying Pauling's electrostatic rule.13 Each μ3-SPh site within the cluster gave a bond valence of 1 when surrounded by two Ag(I) and one Cd(II). Therefore, the four Ag atoms were present in the core of the cluster in three possible arrangements (Fig. S2, ESI†). The Cd atoms were located in the two residual internal sites and the four terminal metal sites. This arrangement of metal atoms in the cluster was consistent with those of several other heterometallic SCCs that have metal atoms/ions of low valence deep within their structures.14–16 The structure of the crystallized cluster 1 was solved with one possible arrangement for the inner Cd and Ag atoms (Fig. 1a and b). The microporous framework 2 crystallized in the tetragonal space group I4(1)/a. In the structure of the microporous framework 2, each cluster 1 was ligated four times via coordination of the terminal Cd atoms by the nitrogen donors of the rigid bpe ligands. The microporous framework 2 had a three-dimensional structure with a diamond topology that belonged to the dia net.17Fig. 1d shows five interpenetrated single frameworks within two layers in 2. Such interpenetrated arrangements are common in MOFs and covalent organic frameworks, particularly those with diamond topologies.10,18,19
:Ag:S atomic ratio of 6.4:4:16.2 (Fig. S1, ESI†). Cd(II) and Ag(I) are isoelectronic; therefore, their relative positions within the clusters cannot be determined by X-ray diffraction. Their positions were instead inferred by applying Pauling's electrostatic rule.13 Each μ3-SPh site within the cluster gave a bond valence of 1 when surrounded by two Ag(I) and one Cd(II). Therefore, the four Ag atoms were present in the core of the cluster in three possible arrangements (Fig. S2, ESI†). The Cd atoms were located in the two residual internal sites and the four terminal metal sites. This arrangement of metal atoms in the cluster was consistent with those of several other heterometallic SCCs that have metal atoms/ions of low valence deep within their structures.14–16 The structure of the crystallized cluster 1 was solved with one possible arrangement for the inner Cd and Ag atoms (Fig. 1a and b). The microporous framework 2 crystallized in the tetragonal space group I4(1)/a. In the structure of the microporous framework 2, each cluster 1 was ligated four times via coordination of the terminal Cd atoms by the nitrogen donors of the rigid bpe ligands. The microporous framework 2 had a three-dimensional structure with a diamond topology that belonged to the dia net.17Fig. 1d shows five interpenetrated single frameworks within two layers in 2. Such interpenetrated arrangements are common in MOFs and covalent organic frameworks, particularly those with diamond topologies.10,18,19
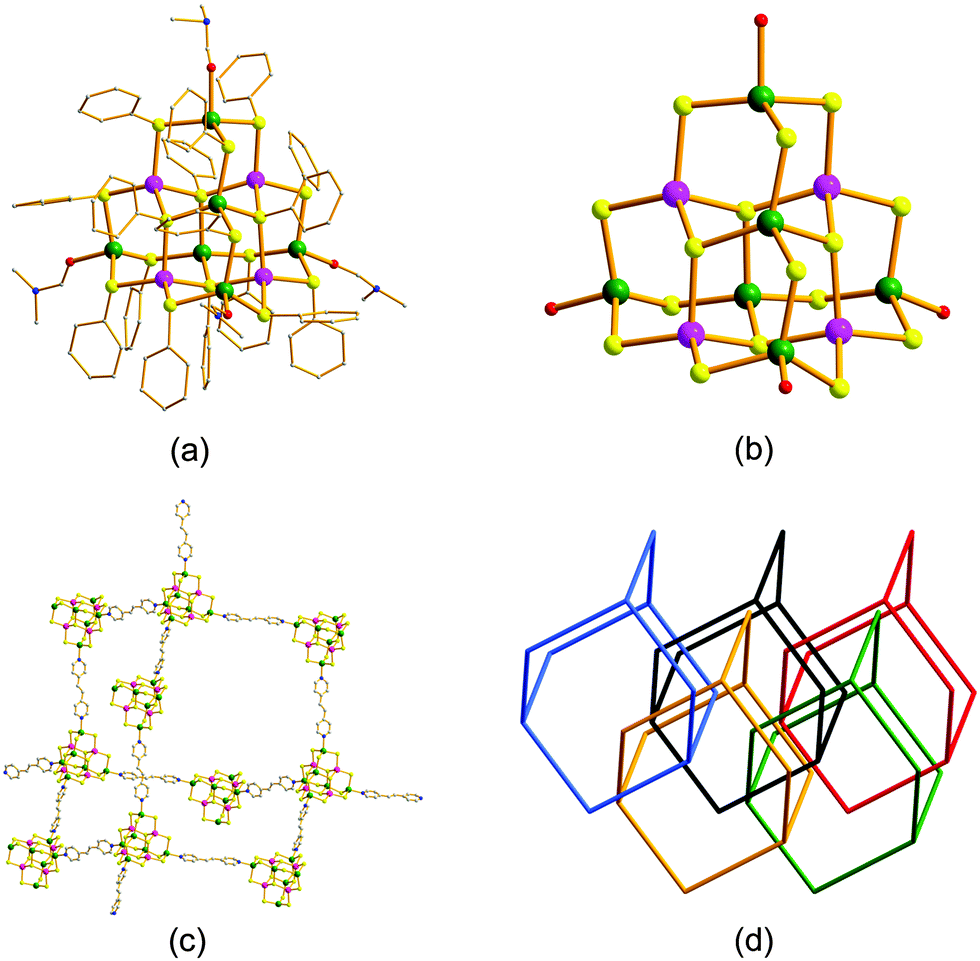 | ||
| Fig. 1 (a) Molecular and (b) core structures of cluster 1 Cd6Ag4(SPh)16(DMF)4 showing one possible arrangement of silver atoms. Hydrogen atoms are omitted for clarity. Green, Cd; pink, Ag; yellow, –SPh; red, oxygen; blue, nitrogen; grey, carbon. (c) Non-interpenetrated visualization of the framework 2 {[Cd6Ag4(SPh)16](bpe)2} based on clusters 1 interlinked by trans-1,2-bis-(4-pyridyl)ethylene (bpe). (d) Actual five-fold interpenetration that occurred in the three-dimensional frameworks of the semiconducting microporous framework 2 based on the clusters and the bpe linkers. | ||
Interpenetration generates small pores that are suitable for enhanced CO2 adsorption from dilute gas mixtures.20 The large voids of 20 × 24 Å in a hypothetical non-interpenetrated version of the framework of 2 became occupied almost fully by the interpenetrations, leaving only very small pores. The porosity was determined by measuring CO2 adsorption. The microporous framework 2 could adsorb CO2 at 11.95 cm3 g−1; its specific surface area (SBET) was moderate (186 m2 g−1 at 1 atm and 273 K). These values are comparable with the somewhat related compound SCIF-3.12 An ultramicroporous network of 6–8 Å pores was calculated for 2 using density functional theory and the CO2 adsorption data (Fig. 2).
 | ||
| Fig. 2 CO2 adsorption isotherm of the microporous semiconducting framework 2 {[Cd6Ag4(SPh)16](bpe)2}n, recorded at 273 K. The inset shows the pore size distribution of 2 derived using density functional theory and the CO2 adsorption isotherm. | ||
Heterometallic clusters are also promising precursors for the preparation of ternary nanocrystals such as CuInS2 and CuInSe2, which are efficient materials for solar cells.21 Interestingly, hybrid crystals of CdS–Ag formed upon the thermal decomposition of cluster 1 at 500 °C in a N2 atmosphere. (A diffraction pattern is shown in Fig. S3, ESI.†) The cluster was thermally stable, showing a decomposition temperature of 320 °C; the microporous framework 2 was less stable due to the bpe (Fig. S6, ESI†). Selective control of the decomposition might allow cluster 1 to produce a ternary CdxAg2−2xS compound.22 Further study of the decomposition of clusters such as 1 might yield new means to prepare hybrid or ternary semiconductors.
The microporous framework 2 showed much greater absorption in the visible range during solid-state UV/vis spectroscopy than did either starting material, Cd(SPh)2 or cluster 1 (Fig. 3). The red shift is interpreted as a combined effect that arose from the proximity of the clusters and their interlinking by bpe. The microporous framework showed broad absorption at wavelengths of 200–600 nm, with a maximum at 305 nm and a shoulder peak at 420 nm. The high-energy absorption peak was assigned to the ternary clusters and the charge transfer from SPh− to Cd2+ or Ag+; the lower-energy shoulder peak was assigned to charge transfer between the conjugated bpe linkers and the clusters. The shoulder absorption peak was reduced in intensity when the framework was dispersed in DMF (not shown). The spectrum of the precursor Cd(SPh)2 was typical for a semiconducting material with a polymeric structure of [Cd4(SPh)8]n. Its absorption onset at 352 nm (3.52 eV) was consistent with the work of Liu et al.23 The absorption spectrum of cluster 1 with its ten metal centers was red-shifted to higher wavelengths compared with the spectrum of the precursor (absorption onset 390 nm, 3.18 eV). The red shift was expected and rationalized by quantum confinement effects.24 The value of the absorption onset for cluster 1 was similar to that of a related compound [Cd10S4(SPh)12] with the same number of d10 centers.25
 | ||
| Fig. 3 Solid-state UV/vis absorption spectra of starting material Cd(SPh)2 (dashed), cluster 1 [Cd6Ag4(SPh)16(DMF)4] (■), and microporous framework 2 {[Cd6Ag4(SPh)16] (bpe)2}n (□). | ||
Given the wide bandgap and the strong visible absorption shown by cluster 1 and microporous framework 2, both are expected to be active photocatalysts. Fig. 4 shows the removal of aqueous rhodamine B (RhB) after illumination. The clusters 1 reduced the dye concentration by 95% after illumination for 180 min. An aqueous dispersion of the microporous framework 2 was an effective photocatalyst; it reduced the concentration of RhB by 95% after illumination for 90 min. The improved photocatalytic activity of the microporous framework might be attributable to its hybrid structure and broad visible absorption, which facilitate the efficient use of incoming energy for the degradation. The photocatalytic performances of both 1 and 2 are comparable to some good photocatalysts for degradation of RhB, such as nanosized Bi2WO6.26 Films of the microporous framework might be better photocatalysts than the dispersed sample due to their further enhanced visible absorption. A blank experiment without any catalyst showed no degradation. The characteristic adsorption band for RhB at 555 nm was chosen for observation of the photocatalytic degradation. The intensity of this band was reduced during successive illumination in the presence of catalysts, and the red solution quickly faded to pale pink (Fig. 4). The degradation of RhB can be explained by either the destruction of its conjugated structure or by deethylation.27 The wavelength of the intensity maximum (λmax) was slightly blue-shifted (from 555 to 529 nm) upon successive illumination, indicating the destruction of the conjugated structure of RhB.
 | ||
| Fig. 4 (a) Decay of RhB concentration calculated from UV/vis absorbance at 555 nm, photocatalyzed by (■) cluster 1 – [Cd6Ag4(SPh)16(DMF)4], (●) microporous framework 2 – {[Cd6Ag4(SPh)16](bpe)2}n, and also (▲) without catalyst; (b) UV/vis spectra and color images showing the decomposition of aqueous rhodamine B (4.2 × 10−5 M) photocatalyzed by the microporous framework 2. | ||
To conclude, a ternary chalcogenolate cluster with a supertetrahedral structure was synthesized and assembled into a solid, porous framework with diamond topology and five-fold interpenetration. The rigid linker bpe aided the formation of a three-dimensional, porous structure, and likely contributed to the emergence of new properties in the framework, leading to a significant red shift of the absorption of the porous framework. Both the cluster 1 and the microporous framework 2 displayed wide band gaps, and were effective photocatalysts under illumination by visible light. We expect that similar clusters and linkers can be crystallized from solutions of DMF into potentially useful structures such as semiconducting films.
This work was supported by the National Science Foundation of China (90922008), the Berzelii Center EXSELENT supported by VR and VINNOVA, and the Swedish Energy Agency. We also thank Bin Qian for optical spectra and Dr Jie Su for crystallographic discussions.
Notes and references
- H. Li, A. Laine, M. O'Keeffe and O. M. Yaghi, Science, 1999, 283, 1145–1147 CrossRef CAS.
- P. Feng, X. Bu and N. Zheng, Acc. Chem. Res., 2005, 38, 293–303 CrossRef CAS PubMed.
- H. Li, J. Kim, T. L. Groy, M. O'Keefe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 4867–4868 CrossRef CAS.
- M. Yaghi, Z. Sun, D. A. Richardson and T. L. Groy, J. Am. Chem. Soc., 1994, 116, 807–808 CrossRef.
- P. Vaqueiro and M. L. Romero, J. Am. Chem. Soc., 2008, 130, 9630–9631 CrossRef CAS PubMed.
- N. Zheng, X. Bu, J. Lauda and P. Feng, Chem. Mater., 2006, 18, 4307–4311 CrossRef CAS.
- J. Xie, X. Bu, N. Zheng and P. Feng, Chem. Commun., 2005, 4916–4918 RSC.
- N. Zheng, X. Bu, H. Vu and P. Feng, Angew. Chem., Int. Ed., 2005, 44, 5299–5303 CrossRef CAS PubMed.
- M. J. Manos, R. G. Iyer, E. Quarez, J. H. Liao and M. G. Kanatzidis, Angew. Chem., Int. Ed., 2005, 44, 3552–3555 CrossRef CAS PubMed.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keefe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- T. Wu, R. Khazhakyan, L. Wang, X. Bu, S.-T. Zheng, V. Chau and P. Feng, Angew. Chem., Int. Ed., 2011, 50, 2536–2539 CrossRef CAS PubMed.
- L. Pauling, J. Am. Chem. Soc., 1929, 51, 1010–1026 CrossRef CAS.
- S. Dehnen and M. K. Brandmayer, J. Am. Chem. Soc., 2003, 125, 6618–6619 CrossRef CAS PubMed.
- W. Su, X. Huang, J. Li and H. Fu, J. Am. Chem. Soc., 2002, 124, 12944–12945 CrossRef CAS PubMed.
- D. F. Back, G. N. M. De Oliveira, R. A. Burrow, E. E. Castellano, U. Abram and E. S. Lang, Inorg. Chem., 2007, 46, 2356–2358 CrossRef CAS PubMed.
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782–1789 CrossRef CAS PubMed.
- B. Kesanli, Y. Cui, M. R. Smith, E. W. Bittner, B. C. Bockrath and W. Lin, Angew. Chem., Int. Ed., 2005, 44, 72–75 CrossRef CAS PubMed.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- S. L. Castro, S. G. Bailey, R. P. Raffaelle, K. K. Banger and A. F. Hepp, Chem. Mater., 2003, 15, 3142–3147 CrossRef CAS.
- J. Chaturvedi, S. Singh, S. Bhattacharya and H. Nöth, Inorg. Chem., 2011, 50, 10056–10069 CrossRef CAS PubMed.
- Y. Liu, F. Boey, L. L. Lao, H. Zhang, X. Liu and Q. Zhang, Chem.–Asian J., 2011, 6, 1004–1006 CrossRef CAS PubMed.
- V. N. Soloviev, A. Eichhöfer, D. Fenske and U. Banin, J. Am. Chem. Soc., 2001, 123, 2354–2364 CrossRef CAS PubMed.
- M. Bendova, M. Puchberger and U. Schubert, Eur. J. Inorg. Chem., 2010, 3299–3306 CrossRef CAS.
- H. Fu, C. Pan, W. Yao and Y. Zhu, J. Phys. Chem. B, 2005, 109, 22432–22439 CrossRef CAS PubMed.
- P. Lei, C. Chen, J. Yang, W. Ma, J. Zhao and L. Zang, Environ. Sci. Technol., 2005, 39, 8466–8474 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis and photocatalysis, XRD patterns, X-ray crystallography data (CIF), additional tables and figures. CCDC 970730 and 970731. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc49660k |
| This journal is © The Royal Society of Chemistry 2014 |