 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxovanadium(V)-induced diastereoselective oxidative homocoupling of boron enolates†
Toru
Amaya
a,
Takaya
Masuda
a,
Yusuke
Maegawa
a and
Toshikazu
Hirao
*ab
aDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Yamada-oka, Suita, Osaka 565-0871, Japan. E-mail: hirao@chem.eng.osaka-u.ac.jp
bJST, ACT-C, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 17th January 2014
Abstract
Oxovanadium(V)-induced dl-selective oxidative coupling of (Z)-boron enolate was demonstrated to give the corresponding 2,3-disubstituted 1,4-diketone in a good yield. High selectivity (up to 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6) was attained when the reaction was performed with VO(OPr-i)2Cl at −30 °C.
:6) was attained when the reaction was performed with VO(OPr-i)2Cl at −30 °C.
Oxidative coupling of enolates can directly afford a 1,4-dicarbonyl skeleton. This is one of the most straightforward approaches to construct such a skeleton, which is often found in natural products, medicinal compounds, organic materials, and their synthetic intermediates. So far, a lot of research has been conducted to develop this type of oxidative coupling reaction.1
The reaction focused on here is intermolecular oxidative homocoupling of ketone enolates. This kind of coupling reaction has been investigated since 1974, and the combination of various oxidants and enolates have been investigated to date.2 However, there is only limited information on the diastereoselective coupling reaction.3 This contrasts sharply with the oxidative homocoupling reaction of acyl enolates, in which the chiral auxiliaries can be utilized.4 As a special case, the enolate of (1R)-camphor was exhibited to undergo the stereoselective oxidative homocoupling.3c Recently, chiral cyclohexenones were revealed to be oxidatively coupled to afford the corresponding 1,4-diketones diastereoselectively.3e As a general strategy, the diastereoselective oxidative coupling of the metalated (S)-(−)-1-amino-2-methoxymethylpyrrolidine (SAMP) or its (R)-isomer, RAMP hydrazone, with iodine was reported,3a where the oxidative cleavage of the N–N bond of the hydrazone using ozone is required to obtain the corresponding ketones, and the overall yields remain low to moderate. Another approach for the diastereoselective synthesis includes the intramolecular oxidative coupling using a traceless tether such as titanium and silicon for two ketone enolates, although the tethered enolate intermediate requires a synthetic step for its preparation.1b,3b,d
Boron enolates have been utilized in a variety of organic syntheses that include a stereoselective aldol reaction.5 Nevertheless, boron enolates have not been used for the oxidative homocoupling reaction to the best of our knowledge. In the present study, boron enolates are focused on from this point of view. So far, we have studied the oxidative carbon–carbon bond formation induced by oxovanadium(V) compounds,6 including the oxidative homo- and cross-coupling of silyl enol ethers to form the corresponding 1,4-diketones.2j Here, we report the oxidative homocoupling of boron enolates using oxovanadium(V) compounds. Notably, a high yield with high dl-selectivity was exhibited.
Boron enolates were prepared via 1,4-hydroboration of enones (Scheme 1).7 This reaction was monitored by 1H NMR spectroscopy to check the conversion and geometric configuration. To a CDCl3 solution of enone 1a the THF solution of 9-borabicyclo[3.3.1]nonane(9-BBN) was added in the presence of MS4A at room temperature. After 2 h, the peaks for enone 1a disappeared in the 1H NMR spectrum. Instead, a characteristic quartet peak appeared at 5.55 ppm, assignable to the vinyl proton in boron enolate 2a. This enolate was formed as a sole diastereomer almost quantitatively. Its stereo-configuration was determined to be (Z)-form based on the observation of a NOE between the ortho-protons of the phenyl group and vinyl proton. To the thus-obtained boron enolate three molar equivalents of VO(OEt)Cl2 were added at room temperature (Scheme 1). After 2 h, the mixture was subjected to aqueous work-up. The desired homocoupled product 3a was obtained in 82% yield, where the diastereomeric ratio (dl/meso) was 67:33.8 Propiophenone (4a) and 2-chloropropiophenone (5a) were observed as side products in this reaction (5% and 13% yields, respectively). The yield of 3a after a 5-minute reaction was comparable to that after 2 h (83%, Table 1, entry 2), indicating that the reaction proceeded fast. Lowering the amount of the oxidant to one molar equivalent caused the yield of 3a to decrease a little (76%, Table 1, entry 3). In this entry, 51V NMR was measured after the reaction. Consequently, the characteristic peak at −294 ppm for VO(OEt)Cl2 disappeared to lead to a vanadium(IV) species (Fig. 1). Further decreasing the amount of the oxidant (0.5 molar equivalents) yielded 3a in 47% yield. These results show that the oxovanadium(V) oxidant formally works as a one-electron oxidant toward the boron enolate.
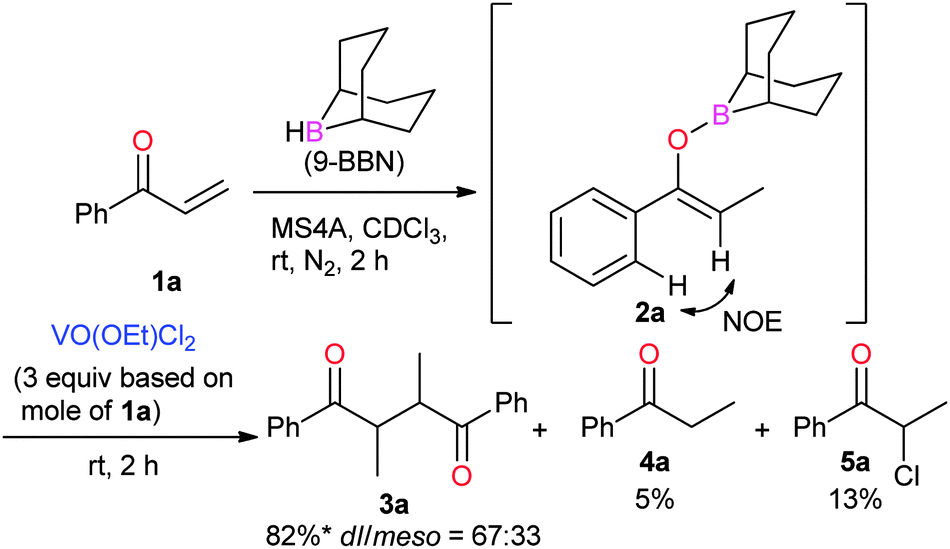 | ||
| Scheme 1 Formation of boron enolate 2a and the oxidative homocoupling with VO(OEt)Cl2. * Yield of 3a = (mole of 3a) × 2/(mole of 1a) × 100. | ||
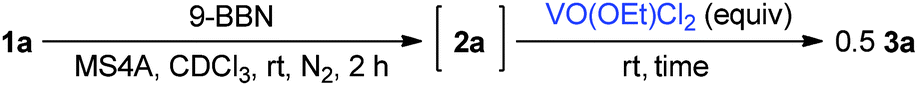
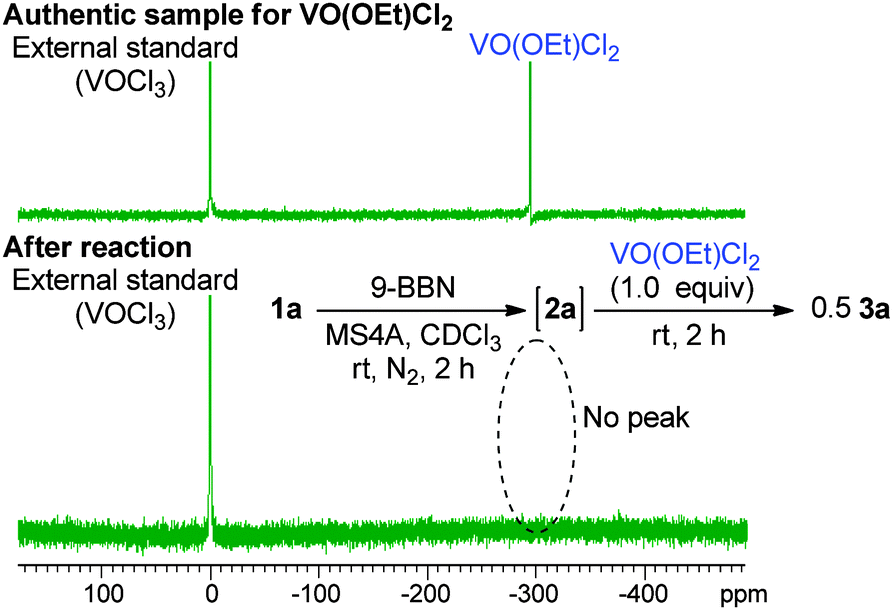 | ||
| Fig. 1 51V NMR spectra of the authentic sample of VO(OEt)Cl2 and after the reaction. | ||
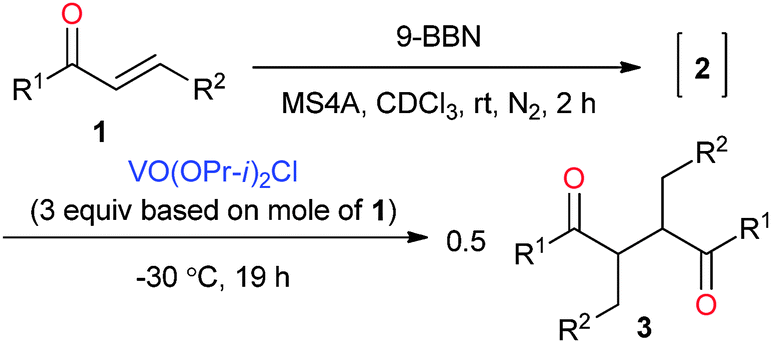
Various oxovanadium(V) oxidants were investigated, as summarised in Table 2. The alkoxy ligands in oxovanadium(V) compounds are known to affect the oxidation reaction. Their oxidation capability usually decreases with the increasing number of alkoxy groups in the complex. Reversely, it increases with the increasing number of the chlorides. The reaction with VO(OPr-i)2Cl resulted in 3a in 78% yield with a dl/meso ratio of 80:20 (Table 2, entry 2), where the diastereoselectivity is higher as compared to that of VO(OEt)Cl2 although the yield is a little lower than that of VO(OEt)Cl2. A lower temperature (−30 °C) increased diastereoselectivity (Table 2, entry 3). Finally, a satisfactory yield and diastereoselectivity was obtained at −30 °C for 19 h (96%, dl/meso = 94:6, Table 2, entry 4), although a scale-up reaction (100 mg scale) slightly decreased the selectivity even at −40 °C (Table 2, entry 5). These reaction conditions were used for the following investigation as optimized conditions. On the other hand, VO(OPr-i)3 did not induce the oxidative coupling due to the lower oxidation capability (Table 2, entry 6). Other oxidants such as FeCl3 and CuCl2 did not induce the oxidative homocoupling (Table 2, entries 7 and 8). In the case of [Ce(NO3)6](NH4)2 (CAN) as an oxidant, 3a was obtained in 53% yield with a low dl/meso selectivity.
|
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Time (h) | Temperature (°C) | Yield of 3ab,c (%), dl/meso ratio |
| a NMR tube experiments. b 1H NMR yield. c Yield = (mole of 3a) × 2/(mole of 1a) × 100. d Same as the result in Scheme 1. e 100 mg scale reaction. f Isolated yield. | ||||
| 1d | VO(OEt)Cl2 | 0.5 | rt | 88, 67:33 |
| 2 | VO(OPr-i)2Cl | 0.5 | rt | 78, 80:20 |
| 3 | VO(OPr-i)2Cl | 0.5 | −30 | 76, 92:8 |
| 4 | VO(OPr-i)2Cl | 19 | −30 | 96, 94:6 |
| 5e | VO(OPr-i)2Cl | 24 | −40 | 93,f 86:14 |
| 6 | VO(OPr-i)3 | 0.5 | rt | Trace |
| 7 | FeCl3 | 19 | −30 | 0 |
| 8 | CuCl2 | 19 | −30 | 0 |
| 9 | CAN | 19 | −30 | 53, 53:47 |

Table 3 shows the scope of the substrates in the oxidative homocoupling using VO(OPr-i)2Cl as an oxidant. All of enones 1 used here were transformed to the corresponding boron enolate stereoselectively. Various p-substitutions of the phenyl group in the boron enolates 1b–f were investigated. Me-, TBSO-, F-, Cl-, and CF3-groups were tolerated without the substitution effect to give the corresponding homocoupling products in high yields with high dl-selectivity (Table 3, entries 1–5).9a–e Instead of the phenyl group, the cyclohexyl group as an example for an aliphatic substrate underwent the homocoupling in 97% yield with a dl/meso selectivity ratio of 77:23 (Table 3, entry 6).9e In the case of (E)-chalcone (1h), the diastereoselectivity was lower although the yield was high (Table 3, entry 7).8




Considering the stereoselectivity based on the steric repulsion between the enolates, the dl-product is favorable in a chelation model and unfavorable in the non-chelation model (see Scheme S1 in the ESI†) as Schmittel and co-workers also discussed the related reaction mechanism in the intramolecular oxidative homocoupling reaction of ketone enolates.3b Oxovanadium alkoxide compounds are known to self-assemble to form a dimer,10 so that the observed dl-selectivity may be accounted for by the intramolecular coupling in the dimer via the formation of the vanadium enolate species. A similar chelation model was also presented in the oxidative homocoupling of ester enolates using TiCl4 to explain the dl-selectivity.11
In conclusion, dl-selective oxidative coupling of (Z)-boron enolate was demonstrated to give the corresponding 2,3-disubstituted 1,4-diketone in a high yield. (Z)-boron enolate was prepared by 1,4-hydroboration of the enone, and it was used for the oxidative coupling reaction in one pot. Stereoselectivity strongly depended on the oxovanadium(V) oxidant and reaction temperature. High selectivity (up to 94:6) was attained when the reaction took place with VO(OPr-i)2Cl at −30 °C. The mechanistic study and synthetic application are now underway.
This work was supported by JST, ACT-C (Japan). Thanks are due to Nichia corporation for the donation of VO(OPr-i)3 and VO(OEt)Cl2.
Notes and references
- For reviews and accounts: (a) A. G. Csákÿ and J. Plumet, Chem. Soc. Rev., 2001, 30, 313–320 RSC; (b) M. Schmittel and A. Haeuseler, J. Organomet. Chem., 2002, 661, 169–179 CrossRef CAS; (c) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (d) F. Guo, M. D. Clift and R. J. Thomson, Eur. J. Org. Chem., 2012, 4881–4896 CrossRef CAS PubMed.
- (a) R. M. Dessau and E. I. Heiba, J. Org. Chem., 1974, 39, 3457–3459 CrossRef CAS; (b) Y. Ito, T. Konoike and T. Saegusa, J. Am. Chem. Soc., 1975, 97, 649–651 CrossRef CAS; (c) Y. Ito, T. Konoike and T. Saegusa, J. Am. Chem. Soc., 1975, 97, 2912–2914 CrossRef CAS; (d) Y. Ito, T. Konoike, T. Harada and T. Saegusa, J. Am. Chem. Soc., 1977, 99, 1487–1493 CrossRef CAS; (e) Y. Kobayashi, T. Taguchi and E. Tokuno, Tetrahedron Lett., 1977, 18, 3741–3742 CrossRef; (f) Y. Kobayashi, T. Taguchi, T. Morikawa, E. Tokuno and S. Sekiguchi, Chem. Pharm. Bull., 1980, 28, 262–267 CrossRef CAS; (g) R. H. Frazier Jr. and R. L. Harlow, J. Org. Chem., 1980, 45, 5408–5411 CrossRef; (h) E. Baciocchi, A. Casu and R. Ruzziconi, Tetrahedron Lett., 1989, 30, 3707–3710 CrossRef CAS; (i) K. Narasaka, T. Okauchi, K. Tanaka and M. Murakami, Chem. Lett., 1992, 2099–2102 CrossRef CAS; (j) T. Fujii, T. Hirao and Y. Ohshiro, Tetrahedron Lett., 1992, 33, 5823–5826 CrossRef CAS; (k) K. Ryter and T. Livinghouse, J. Am. Chem. Soc., 1998, 120, 2658–2659 CrossRef CAS; (l) M. D. Clift, C. N. Taylor and R. J. Thomson, Org. Lett., 2007, 9, 4667–4669 CrossRef CAS PubMed; (m) M. D. Clift and R. J. Thomson, J. Am. Chem. Soc., 2009, 131, 14579–14583 CrossRef CAS PubMed; (n) B. M. Casey and R. A. Flowers II, J. Am. Chem. Soc., 2011, 133, 11492–11495 CrossRef CAS PubMed; (o) Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2012, 48, 5355–5357 RSC; (p) H.-Q. Do, H. Tran-Vu and O. Daugulis, Organometallics, 2012, 31, 7816–7818 CrossRef CAS PubMed.
- (a) D. Enders, P. Müller and D. Klein, Synlett, 1998, 43–44 CrossRef CAS PubMed; (b) M. Schmittel, A. Burghart, W. Malisch, J. Reising and R. Söllner, J. Org. Chem., 1998, 63, 396–400 CrossRef CAS; (c) J. McNulty, M. J. Millar, G. Bernardinelli and C. W. Jefford, J. Org. Chem., 1999, 64, 5312–5314 CrossRef CAS; (d) C. T. Avetta Jr., L. C. Konkol, C. N. Taylor, K. C. Dugan, C. L. Stern and R. J. Thomson, Org. Lett., 2008, 10, 5621–5624 CrossRef PubMed; (e) F. Guo, L. C. Konkol and R. J. Thomson, J. Am. Chem. Soc., 2011, 133, 18–20 CrossRef CAS PubMed.
- (a) N. A. Porter, Q. Su, J. J. Harp, I. J. Rosenstein and A. T. McPhail, Tetrahedron Lett., 1993, 34, 4457–4460 CrossRef CAS; (b) N. Kise, K. Tokioka, Y. Aoyama and Y. Matsumura, J. Org. Chem., 1995, 60, 1100–1101 CrossRef CAS; (c) T. Langer, M. Illich and G. Helmchen, Tetrahedron Lett., 1995, 36, 4409–4412 CrossRef CAS; (d) T. Langer, M. Illich and G. Helmchen, Synlett, 1996, 1137–1139 CrossRef CAS PubMed; (e) N. Kise, K. Kumada, Y. Terao and N. Ueda, Tetrahedron, 1998, 54, 2697–2708 CrossRef CAS.
- (a) B. M. Kim, S. F. Williams and S. Masamune, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, New York, 1991, vol. 2, pp. 239–275 Search PubMed; (b) C. J. Cowden and I. Paterson, Org. React., 1997, 51, 1–200 CAS; (c) T. Mukaiyama and J.-I. Matsuo, in Modern Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Weinheim, Germany, 2004, ch. 3, vol. 1 Search PubMed.
- (a) T. Hirao, T. Fujii and Y. Ohshiro, Tetrahedron, 1994, 50, 10207–10214 CrossRef CAS; (b) T. Ishikawa, A. Ogawa and T. Hirao, Organometallics, 1998, 17, 5713–5716 CrossRef CAS; (c) T. Ishikawa, A. Ogawa and T. Hirao, J. Am. Chem. Soc., 1998, 120, 5124–5125 CrossRef CAS; (d) T. Ishikawa, S. Nonaka, A. Ogawa and T. Hirao, Chem. Commun., 1998, 1209–1210 RSC; (e) T. Ishikawa, A. Ogawa and T. Hirao, J. Organomet. Chem., 1999, 575, 76–79 CrossRef CAS; (f) T. Hirao, T. Takada and A. Ogawa, J. Org. Chem., 2000, 65, 1511–1515 CrossRef CAS PubMed; (g) T. Hirao, T. Takada and H. Sakurai, Org. Lett., 2000, 2, 3659–3661 CrossRef CAS PubMed; (h) T. Takada, H. Sakurai and T. Hirao, J. Org. Chem., 2001, 66, 300–302 CrossRef CAS; (i) H. Mizuno, H. Sakurai, T. Amaya and T. Hirao, Chem. Commun., 2006, 5042–5044 RSC; (j) T. Amaya, Y. Tsukamura and T. Hirao, Adv. Synth. Catal., 2009, 351, 1025–1028 CrossRef CAS.
- Y. Matsumoto and T. Hayashi, Synlett, 1991, 349–350 CAS.
- The diastereomeric ratio (dl/meso) was determined by 1H NMR. The assignment for dl and meso compounds was according to the following reference: S. E. Drewes, C. J. Hogan, P. T. Kaye and G. H. P. Roos, J. Chem. Soc., Perkin Trans. 1, 1989, 1585–1591 RSC.
- The diastereomeric ratio (dl/meso) was determined by 1H NMR. The assignment for dl and meso compounds was performed as follows: (a) For 3b: H. Ikeda, T. Takasaki, Y. Takahashi, A. Konno, M. Matsumoto, Y. Hoshi, T. Aoki, T. Suzuki, J. L. Goodman and T. Miyashi, J. Org. Chem., 1999, 64, 1640–1649 CrossRef CAS PubMed; (b) For 3c: S. G. A. Moinuddin, S. Hishiyama, M.-H. Cho, L. B. Davin and N. G. Lewis, Org. Biomol. Chem., 2003, 1, 2307–2313 RSC; (c) For 3d: the dl-isomer was determined with X-ray crystallographic analysis (CCDC-969201), see ESI†; (d) For 3e: the dl-isomer was determined with X-ray crystallographic analysis (CCDC-969200), see ESI†; (e) For 3f and 3g: although the assignments could not be confirmed, they are predicted according to the tendency of 1H NMR spectra for 3b–3e, and 3h.
- C. N. Caughlan, H. M. Smith and K. Watenpaugh, Inorg. Chem., 1966, 5, 2131–2134 CrossRef CAS.
- Y. Matsumura, M. Nishimura, H. Hiu, M. Watanabe and N. Kise, J. Org. Chem., 1996, 61, 2809–2812 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, and compounds characterization, including 1H and 13C NMR spectra, and X-ray crystallographic analysis for dl-3d and dl-3e. CCDC 969200 and 969201. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc49624d |
| This journal is © The Royal Society of Chemistry 2014 |