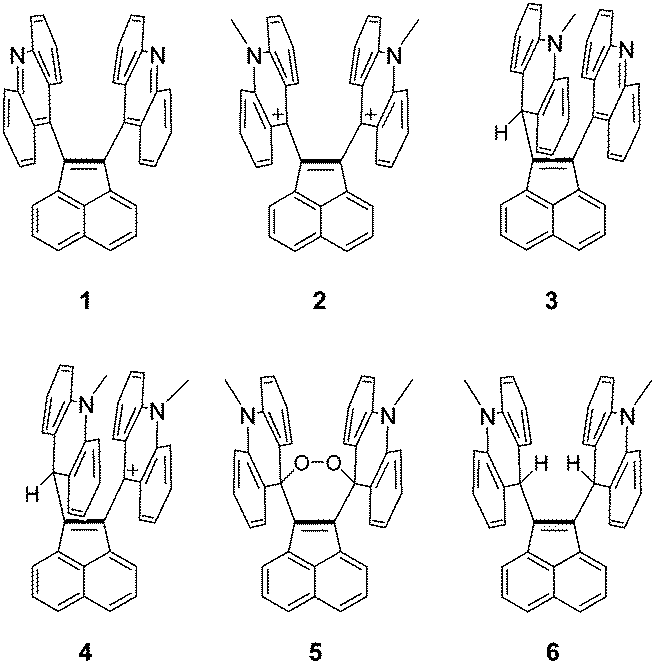
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation and structure of acenaphthylene-1,2-diyldi(9-acridine) derivatives with a long C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C bond†
C bond†
Takashi
Takeda
*a,
Yasuto
Uchimura
b,
Hidetoshi
Kawai
c,
Ryo
Katoono
b,
Kenshu
Fujiwara
b and
Takanori
Suzuki
*b
aInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Sendai, Miyagi 980-8577, Japan. E-mail: takeda@tagen.tohoku.ac.jp; Fax: +81-22-217-5654; Tel: +81-22-217-5655
bDepartment of Chemistry, Faculty of Science, Hokkaido University, Sapporo, Hokkaido 060-0810, Japan. E-mail: tak@mail.sci.hokudai.ac.jp; Fax: +81-11-706-2714; Tel: +81-11-706-2714
cDepartment of Chemistry, Faculty of Science, Tokyo University of Science, Shinjuku, Tokyo 162-8601, Japan
First published on 20th January 2014
Abstract
Due to purely steric effects of acridine units, acenaphthylene-1,2-diyldi(9-acridine) has a long C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond [1.3789(19) Å] while maintaining its sp2 hybridized nature and bond order.
C bond [1.3789(19) Å] while maintaining its sp2 hybridized nature and bond order.
It is important to achieve a better understanding of the nature of covalent bonds because covalent bonding is a fundamental concept in chemistry. The comparison of “normal” bonds to bonds with unusual parameters, such as bond lengths or angles, is an important method for gaining insight into covalent bonds. Many studies have been based on this idea,1 and especially on the C–C single bond, which is one of the most common covalent bonds in organic compounds. Long C–C bonds have been found in hexaphenylethane derivatives,2 in which steric repulsion between six aryl groups around the C–C bond causes bond elongation.3 Extreme examples have been found in derivatives of tetraarylbenzocyclobutene2b,c and tetraarylpyracene,2d–f in which the C–C bond is longer than 1.7 Å (standard value: 1.54 Å).
Strained CC bonds have been used as highly reactive intermediates in synthetic chemistry.4 There are several types of strained CC bonds,5,6 including CC bonds with a twisted structure6 or pyramidalized carbons, an unusual CC–C bond angle, and a long or short bond length (Fig. 1).
 | ||
| Fig. 1 Types of strain in CC bonds. | ||
As in the case of highly expanded C–C bonds, the elongation of a CC double bond would be induced by geometrical strain. However, there have been fewer successful studies on the elongation of a CC double bond while maintaining its sp2-hybridized nature and bond order. The major concern is that the π-bond order of the CC bond is easily reduced just by the attachment of electron-donating/-accepting substituents or π-systems. Therefore, in many cases, the observed bond elongation resulted from the reduced bond order but not from geometrical strain. Furthermore, it has been reported that the CC bond can be stretched by the introduction of cationic coordinating substituents adjacent to the CC bond: e.g., the coordination of cationic transition metals to 1,2-diaminoethylene causes CC bond elongation.7 However, this method also decreases the π-bond order because of the formation of carbon–metal bonds. To investigate the relationship between the bond length and the nature of the CC double bond, we must exclude such “impure” CC double bonds with the reduced bond order from the following discussion.
Based on studies on the long C–C single bonds in hexaphenylethane derivatives, we expected that a steric effect around two carbon nuclei could be a good way to expand a CC double bond without changing its π-bond order. Since there can be fewer possible substituents around a CC bond than around a C–C bond, expansion of a CC bond by steric repulsion should give a smaller change in length. Thus, precise determination with small experimental errors is essential. Hitherto, there have been few attempts to elongate a CC bond using this approach. It has been reported that, even with the attachment of two bulky groups, the CC bond length in (Z)-1,2-di(1-adamantyl)ethylene8 [1.34(1) Å] is almost the same as the standard CC bond length (1.33 Å),9,10 and the value was determined only with a large estimated standard deviation (0.01 Å). Although octachlorobifluorene has a long CC bond [1.392(4) Å], its elongation is not mainly due to steric repulsion but accompanied by bond twisting (twist angle of 66°) to reduce the π-bond order.6b However, we envisaged that selection of the rigid double-bond scaffold and bulky substituents could still give a chance to expand a CC double bond while maintaining its sp2-hybridized nature and bond order.
As a rational molecular design to attain severe steric repulsion around the CC bond, we selected 1,2-disubstituted acenaphthylenes. Due to the rigid molecular framework of acenaphthylenes, the substituents at the C1 and C2 positions should be placed in close proximity, thus generating enough repulsion to expand the CC bond. Another merit of the use of the acenaphthylene-1,2-diyl skeleton is that the C1C2 bond in a five-membered ring would be expanded before the substituents are attached at the C1 and C2 positions due to the “clamping” effect, which makes it easy for the bond length to be affected by bulky substituents because the prestrained bond is more susceptible to steric perturbation.11
The C1C2 bond length (d1) of parent acenaphthylene estimated by DFT calculations (B3LYP/6-31G*) is 1.364 Å.12 The expansion comes with the “clamping” effect but not due to formal conjugation with the naphthalene subunit. The isolation of the C1C2 bond from conjugation is indicated by the lack of closed-shell resonance structures of acenaphthylene with a C1–C2 single bond. The bond length of C1(or 2)–C8a(or 2a) of acenaphthylene (d2: 1.473 Å, DFT) is almost the same as that of the standard Csp2–Csp2 single bond (1.478 Å for an unconjugated CC–CC bond),10 which supports the above idea. Experimentally determined d1 and d2 values are 1.351(3) and 1.483(3) Å in acenaphthylene-5,6-diyldi(9-acridine),2d and 1.362(3) and 1.488(3)–1.491(2) Å in acefluoranthylene,14 respectively, which is close to the calculated values (1.361 and 1.470 Å for the former and 1.369 and 1.485 Å for the latter, respectively). Thus, the C1C2 bond in acenaphthylenes can be considered to be a prestrained but “pure” CC bond, whose geometries are close to those estimated by DFT calculation.12 Herein, we sought to expand the prestrained C1C2 bond of acenaphthylene by applying purely strain effects to observe the elongated CC bond while maintaining its sp2-hybridized nature and order.
To achieve sufficient steric repulsion around the CC bond to expand the bond, we expected that the introduction of large planar groups would be effective. Because of nearly perpendicular arrangement of the planar groups to the π-plane of the CC bond, the decrease in steric repulsion by skewing of the framework or the pyramidalization of Csp2-carbons is inefficient. Thus, steric repulsion could be relieved only by bond expansion and not by other modes of deformation. Even when we choose planar π-systems as substituents on C1 and C2, their conjugating effects would be negligible due to their twisted arrangement toward the C1C2 bond. Therefore, we designed acenaphthylene-1,2-diyldi(9-acridine) 1 as a promising target for elongating the CC bond without changing its sp2-hybridized nature and bond order. The acridine substituent in 1 is more feasible than a hydrocarbon counterpart due to its modifiability into several derivatives 2–6, the structures of which offer useful information for discussing the steric/electronic effects on bond elongation in 1. The details will be described below.
Acenaphthylene-1,2-diyldiacridine 1 and its derivatives 2–6 were prepared as summarized in Scheme 1. The CuO-promoted Stille coupling15 of 1,2-dibromoacenaphthylene 716 with 9-(trimethylstannyl)acridine 817 gave diacridine 1 as a sparingly soluble yellow solid. Dimethylation by MeOTf in CH2Cl2 gave dication salt 2·(TfO−)2. To prevent formation of monomethyl-monoprotonated species as by-products, addition of a hindered base (2,6-di-tert-butyl-4-methylpyridine) was necessary. The mixture of 2·(TfO−)2 and the pyridinium salt was treated with NaBH4 to give bis(acridan) 6. Oxidation of 6 with (p-BrC6H4)3NSbCl6 regenerated dication 2 as the SbCl6− salt. On the other hand, diacridine 1 was selectively monomethylated by MeOTf in benzene, and the resulting monocation was treated with NaBH4 to give the unsymmetric acridan–acridine hybrid 3, which was further methylated to give the acridan–acridinium complex 4·TfO−. Selective monomethylation in benzene could be accounted for by insolubility of the resulting monocation salt in this solvent. We unexpectedly found that, under aerated alkaline conditions, 4·TfO− can be transformed into peroxide 5. Although the precise reaction mechanism of formation of peroxide is still unclear, it might include deprotonation of the methine proton of acridan to form neutral acenaphthenequinodimethane 9 followed by reaction with oxygen. Peroxide formation from an electron-donating tetraarylacenaphthenequinodimethane was previously reported by our group.18
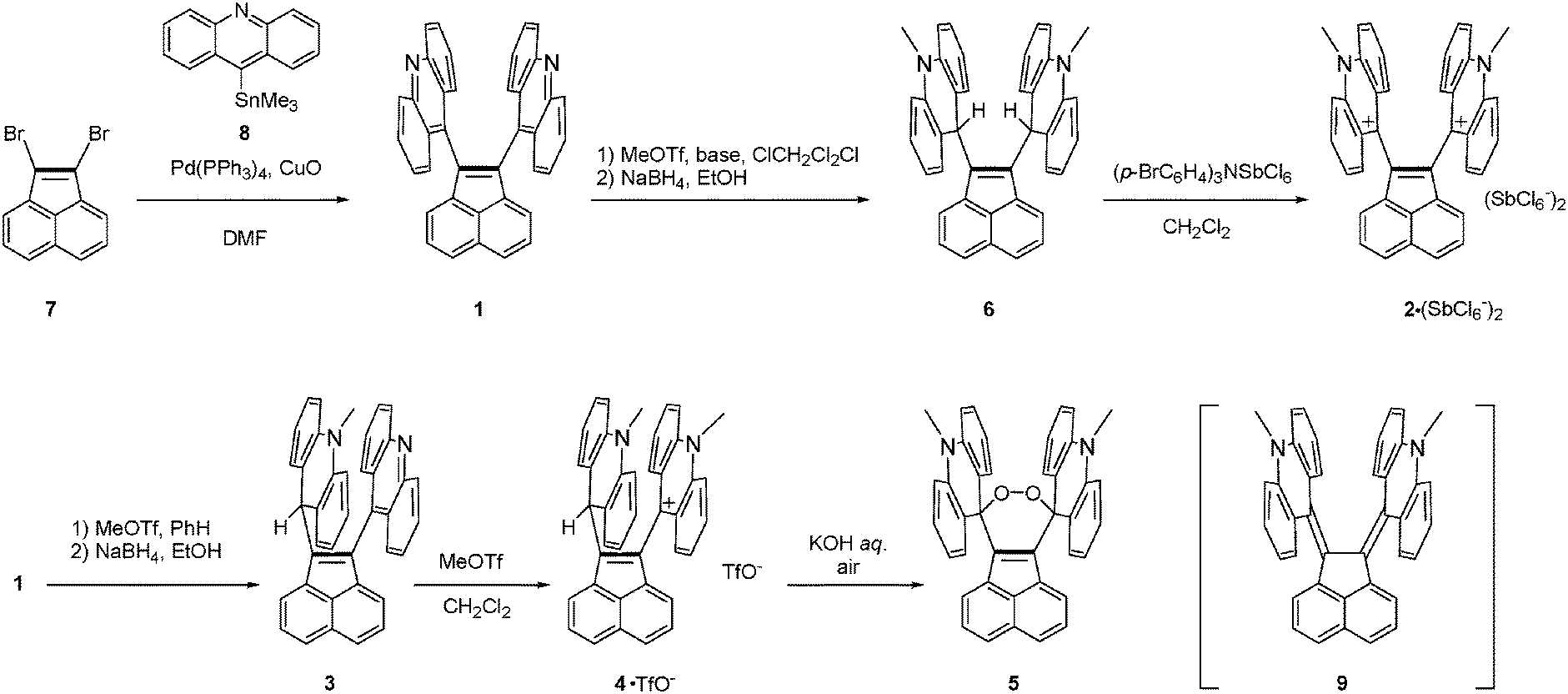 | ||
| Scheme 1 Preparation of acenaphthylene-1,2-diyldiacridine 1 and its derivatives 2–6. | ||
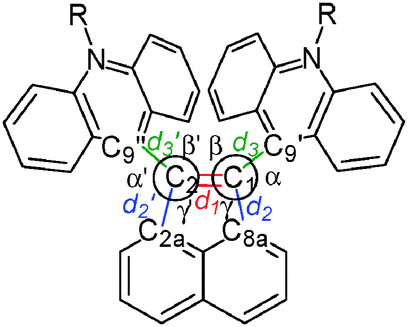
Fig. 2 shows an ORTEP drawing of 1 obtained by low-temperature single-crystal X-ray analyses. Selected structural parameters around the C1C2 bond are summarized in Table 1. The precisely determined C1C2 bond length in 1 [1.3789(19) Å] is clearly much greater than the standard value (1.33 Å) and that in the parent acenaphthylene. Notably, the C1C2 bond in 1 is longer than that in 1,2-di-tert-butyl-3,3,5,5-tetramethylcyclopentene [1.365(2) Å], which is the sole successful example of a long CC bond without disturbing the sp2-hybridized nature or π-bond order.19 As shown by the space-filling model of 1 (Fig. 2), the acridine π units are in strong contact with each other in a parallel fashion. Especially, the C9′⋯C9′′ distance (3.19 Å) is much shorter than the sum of the van der Waals radii (3.40 Å). The torsion angles between acenaphthylene and the two acridines (62.7° and 64.0°, respectively) are large enough to neglect π-conjugation between the acridine units and the C1C2 bond. The C1C2 bond in 1 is significantly longer than the C1C2 bond length in 1,2-di(1-naphthyl)acenaphthylene [1.367(2) Å],20 in which the naphthalene units are arranged in an anti-parallel fashion to reduce steric repulsion. Thus, very severe steric repulsion should be the main contributor to elongation of the C1C2 bond. As designed, different modes of deformation, such as twisting or pyramidalization, seldom occur in 1, and thus the C1C2 unit is almost planar, as shown by the small torsion angles of C9′–C1–C2–C9′′ (X) and C8a–C1–C2–C2a (Y) and by the fact that the sums of the bond angles around C1 and C2 (α + β + γ) are 360° (see Table 1).
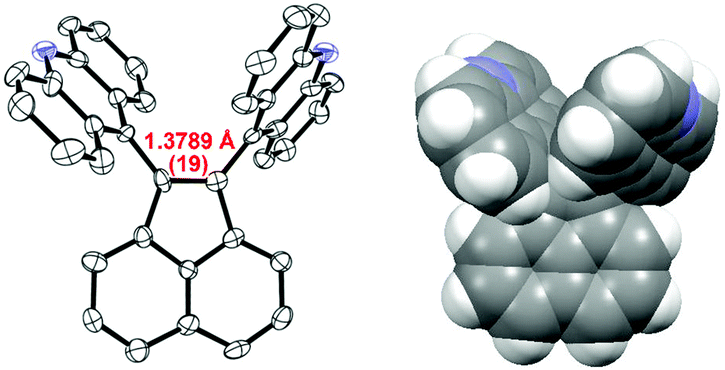 | ||
| Fig. 2 ORTEP drawing (left) and the space-filling model of 1. | ||
C2 bond determined by low-temperature X-ray analyses or DFT optimizations
| Compd | d 1 | d 2 | d 3 | d 2′ | d 3′ | α + β + γ | α′ + β′ + γ′ | X | Y | C9′⋯C9′′ | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| X: torsion angle of C9′–C1–C2–C9′′; Y: torsion angle of C8a–C1–C2–C2a. | |||||||||||
| 1 | 1.3789(19) | 1.474(3) | 1.489(3) | 1.474(3) | 1.489(3) | 359.94(16) | 359.94(16) | 6.3(4) | 1.0(3) | 3.189(3) |
|
| 1·CHCl3 | 1.377(3) | 1.479(3) | 1.478(3) | 1.481(3) | 1.487(3) | 359.98(17) | 360.00(18) | 1.8(4) | 0.63(19) | 3.249(3) | |
| 1 (calc) | 1.383 | 1.482 | 1.485 | 1.482 | 1.485 | 360.0 | 360.0 | 0.34 | 1.01 | 3.232 | |
| 2·MeCN | 1.371(5) | 1.475(4) | 1.480(5) | 1.480(4) | 1.479(4) | 359.9(3) | 359.9(3) | 4.4(4) | 1.8(3) | 3.115(5) | |
| 2·CHCl3 | 1.391(12) | 1.499(10) | 1.464(10) | 1.482(9) | 1.484(12) | 359.9(8) | 359.9(8) | 6.8(10) | 0.4(6) | 3.172(13) | |
| 2 (calc) | 1.397 | 1.474 | 1.481 | 1.474 | 1.482 | 360.0 | 360.0 | 0.70 | 1.53 | 3.268 | |
| 3 | 1.369(4) | 1.474(5) | 1.480(4) | 1.479(4) | 1.525(5) | 360.0(3) | 360.0(3) | 3.9(5) | 0.3(3) | 3.259(4) | |
| 3 (calc) | 1.377 | 1.482 | 1.487 | 1.484 | 1.527 | 360.0 | 360.0 | 0.62 | 0.06 | 3.342 | |
| 4 (mol 1) | 1.374(10) | 1.488(10) | 1.464(10) | 1.477(10) | 1.528(10) | 359.9 (6) | 360.0(6) | 1.5(13) | 1.6(8) | 3.288(10) | |
| (mol 2) | 1.357(10) | 1.503(10) | 1.489(9) | 1.466(10) | 1.504(10) | 360.0(6) | 360.0(6) | 1.6(13) | 1.7(8) | 3.262(9) | |
| 4 (calc) | 1.380 | 1.482 | 1.478 | 1.479 | 1.526 | 360.0 | 360.0 | 0.61 | 1.04 | 3.300 | |
| 5 | 1.356(5) | 1.478(6) | 1.501(6) | 1.477(6) | 1.505(6) | 360.0(4) | 359.8(4) | 8.4(5) | 2.7(4) | 3.015(4) | |
| 5 (calc) | 1.369 | 1.477 | 1.509 | 1.477 | 1.509 | 360.0 | 360.0 | 5.04 | 2.46 | 2.988 | |
| 6 | 1.364(4) | 1.479(3) | 1.500(3) | 1.486(3) | 1.511(4) | 360.0(2) | 360.0(2) | 6.7(5) | 2.3(4) | 3.175(4) | |
| 6 (calc) | 1.377 | 1.483 | 1.510 | 1.484 | 1.521 | 360.0 | 360.0 | 0.00 | 0.00 | 3.207 | |
The C1C2 bond length [1.377(3) Å] and other structural parameters of 1 in the CHCl3 solvate crystal are similar to those of the nonsolvated crystal of 1 shown above (Table 1) despite the difference in the nature of their packing. Thus, the molecular geometry with the elongated C1C2 bond is intrinsic to 1 rather than forced by crystal packing or other intermolecular factors.
To further confirm that π-conjugation is not responsible for elongation of the C1C2 bond in 1, the length of the C1C2 bond was compared to those in its derivatives 2–6. The electron-withdrawing nature of acridiniums in 2 is much higher than that of neutral acridines in 1. If the electronic contribution from the substituents attached to the CC bond is the major factor, the C1C2 bond length of dication 2 should be different from that of 1. However, the C1C2 bond lengths of the two pseudopolymorphs of 2 [1.391(12) Å for the CHCl3 solvate crystal; 1.371(5) Å for the MeCN solvate crystal] determined by low-temperature X-ray analyses were nearly the same as that of 1, as were other structural parameters (Table 1), which can be rationalized by considering that acridinium and acridine have a similar steric bulkiness around the C9-position. Thus, the contribution of an electronic effect to elongation of the C1C2 bond should be negligible.
In addition, further evidence of the absence of conjugation effects could be obtained by comparison of the C1C2 bond lengths of 1 and 2 with those of push–pull type compounds 3 and 4, in which one of the two acridine/acridinium units is replaced by an electron-donating acridan unit. In general, push–pull substitution over the CC bond causes a drastic decrease in the π-bond order, and thus the bond length must be much longer in the presence of conjugation effects between the substituents over C1C2, especially in 4, in which the acridinium is more strongly electron-accepting than the acridine in 3. However, the observed C1C2 bond lengths of 3 [1.369(4) Å] and 4 [1.374(10) Å and 1.357(10) Å; two independent molecules] are similar or rather shorter than those in diacridine 1 and diacridinium 2, which clearly excludes the presence of conjugation effects in the present system. The notable difference in d3′ can be explained by the fact that the standard Csp2–Csp3 single bond is longer than the Csp2–Csp2 single bond.
In the case of peroxide 5 and diacridan 6, steric repulsion between two methylacridan units was obviously relieved by the insertion of two oxygen or hydrogen atoms between the bulky methylacridan units. The C1C2 bond lengths [1.356(5) for 5 and 1.364(4) Å for 6, respectively] became shorter than that in diacridine 1, and are almost the same as that in nonsubstituted acenaphthylene. Based on all of these experimental results, we can safely conclude that steric repulsion is the dominant factor in the elongation of the CC bond in 1. The structures of 1–6 optimized by DFT calculations (B3LYP/6-31G*) well reproduced those determined experimentally by X-ray analyses, which supports the steric effect for the elongation of the C1C2 bond in 1.
In summary, we have demonstrated that steric repulsion in 1 effectively elongated the pure CC double bond by a rational molecular design, in which we could rule out the influence of a π-conjugating electronic effect. The key to success was the use of bulky planar groups to cause severe steric repulsion which could only be relieved by bond elongation, and not by other modes of deformation. Diacridine 1 has a long CC bond of up to 1.3798(19) Å, which is much longer than the standard value. The exclusion of both the conjugation effect and structural deformation guarantees the sp2-hybridized nature and the lack of electronic perturbation for these pure CC double bonds. As molecules with unusual CC bonds are developed, their characteristic physical and electronic properties (e.g., small force constant and long wave absorption involving a long CC bond) and unique reactivities will be investigated. Studies along these lines will be made in due course.
This work was supported in part by Grant-in-Aid for Young Scientists (No. 24750032) and Grant-in-Aid for Scientific Research on Innovative Areas: “Organic Synthesis Based on Reaction Integration” (No. 2105) from MEXT, Japan.
Notes and references
- (a) H. Kawai, T. Suzuki, M. Ohkita and T. Tsuji, Angew. Chem., Int. Ed., 1998, 37, 817–819 CrossRef CAS; (b) L. W. Jenneskens, F. J. J. de Kanter, P. A. Kraakman, L. A. M. Turkenburg, W. E. Koolhaas, W. H. de Wolf, F. Bickelhaupt, Y. Tobe, K. Kakiuchi and Y. Odaira, J. Am. Chem. Soc., 1985, 107, 3716–3717 CrossRef CAS; (c) M. Tanaka and A. Sekiguchi, Angew. Chem., Int. Ed., 2005, 44, 5821–5823 CrossRef CAS PubMed.
- Representative publications (a) J. M. McBride, Tetrahedron, 1974, 30, 2009–2022 CrossRef CAS; (b) F. Toda, Eur. J. Org. Chem., 2000, 1377–1386 CrossRef CAS and references cited therein; (c) S. Kammermeier, P. G. Jones and R. Herges, Angew. Chem., Int. Ed. Engl., 1997, 36, 1757–1760 CrossRef CAS; (d) H. Kawai, T. Takeda, K. Fujiwara, M. Wakeshima, Y. Hinatsu and T. Suzuki, Chem.–Eur. J., 2008, 14, 5780–5793 CrossRef CAS PubMed; (e) T. Takeda, H. Kawai, R. Herges, E. Muche, Y. Sawai, K. Murakoshi, K. Fujiwara and T. Suzuki, Tetrahedron Lett., 2009, 50, 3693–3697 CrossRef CAS PubMed; (f) T. Suzuki, T. Takeda, H. Kawai and K. Fujiwara, Pure Appl. Chem., 2008, 80, 547–553 CrossRef CAS; (g) T. Takeda, Y. Uchimura, H. Kawai, R. Katoono, K. Fujiwara and T. Suzuki, Chem. Lett., 2013, 42, 954–962 CrossRef CAS and references cited therein.
- T. Suzuki, K. Ono, J. Nishida, H. Takahashi and T. Tsuji, J. Org. Chem., 2000, 65, 4944–4948 CrossRef CAS PubMed.
- Review M. R. Wilson and R. E. Taylor, Angew. Chem., Int. Ed., 2013, 52, 4078–4087 CrossRef CAS PubMed.
- Review (a) P. M. Warner, Chem. Rev., 1989, 89, 1067–1093 CrossRef CAS and reference cited therein; (b) D. Lenoir, C. Wattenbach and J. F. Liebman, Struct. Chem., 2006, 17, 419–422 CrossRef CAS.
- (a) A. Beck, R. Gompper, K. Polborn and H.-U. Wagner, Angew. Chem., Int. Ed. Engl., 1993, 32, 1352–1354 CrossRef; (b) E. Molins, C. Miravitlles, E. Espinosa and M. Ballester, J. Org. Chem., 2002, 67, 7175–7178 CrossRef CAS PubMed.
- For examples of complex of 1,2-diaminoacenaphthylenes having long C1C2 bonds see:
(a) H. Tsurugi, T. Saito, H. Tanahashi, J. Arnold and K. Mashima, J. Am. Chem. Soc., 2011, 133, 18673–18683 CrossRef CAS PubMed;
(b) I. L. Fedushkin, V. A. Chudakova, A. A. Skatova, N. M. Khvoinova, Y. A. Kurskii, T. A. Glukhova, G. K. Fukin, S. Dechert, M. Hummert and H. Schumann, Z. Anorg. Allg. Chem., 2004, 630, 501–507 CrossRef CAS;
(c) I. L. Fedushkin, N. M. Khvoinova, A. Yu. Baurin, G. K. Fukin, V. K. Cherkasov and M. P. Bubnov, Inorg. Chem., 2004, 43, 7807–7815 CrossRef CAS PubMed;
(d) A. Paulovicova, U. El-Ayaan, K. Umezawa, C. Vithana, Y. Ohashi and Y. Fukuda, Inorg. Chim. Acta, 2002, 339, 209–214 CrossRef CAS;
(e) I. L. Fedushkin, O. V. Maslova, M. Hummert and H. Schumann, Inorg. Chem., 2010, 49, 2901–2910 CrossRef CAS PubMed.
- A. P. Marchand, D. Xing and S. G. Bott, Tetrahedron Lett., 1994, 35, 8935–8938 CrossRef CAS.
- L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystal: An Introduction to Modern Structural Chemistry, Cornell University Press, Ithaca, NY, 3rd edn, 1960 Search PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- For example, see: (a) T. Suzuki, Y. Uchimura, Y. Ishigaki, T. Takeda, R. Katoono, H. Kawai, K. Fujiwara, A. Nagaki and J. Yoshida, Chem. Lett., 2012, 41, 541–543 CrossRef CAS; (b) K. Wada, T. Takeda, H. Kawai, R. Katoono, K. Fujiwara and T. Suzuki, Chem. Lett., 2013, 42, 1194–1196 CrossRef CAS.
- Due to notorious positional disorder, in-plane rotational motion, and phase transition of the parent acenaphthene crystal (ref. 13), the diffraction experiments gave somewhat strange d1 value [1.395(11) Å], which might be related to the rigid-body structural refinement by assuming a higher molecular symmetry than the actual one in the crystal for the three crystallographically independent molecules.
- (a) R. A. Wood, T. R. Welberry and A. D. Rae, J. Chem. Soc., Perkin Trans. 2, 1985, 451–456 RSC; (b) T. R. Welberry, Proc. R. Soc. London, Ser. A, 1973, 334, 19–48 CrossRef CAS.
- M. Lutz, A. L. Spek, M. Sarobe and L. W. Jenneskens, Acta Crystallogr., Sect. C, 1999, 55, 659–661 Search PubMed.
- (a) C. Wolf and X. Mei, J. Am. Chem. Soc., 2003, 125, 10651–10658 CrossRef CAS PubMed; (b) X. Mei and C. Wolf, J. Org. Chem., 2005, 70, 2299–2305 CrossRef CAS PubMed; (c) X. Mei, R. M. Martin and C. Wolf, J. Org. Chem., 2006, 71, 2854–2861 CrossRef CAS PubMed.
- B. M. Trost and D. R. Brittelli, J. Org. Chem., 1967, 32, 2620–2621 CrossRef CAS.
- (a) H. Kawai, T. Takeda, K. Fujiwara and T. Suzuki, J. Am. Chem. Soc., 2005, 127, 12172–12173 CrossRef CAS PubMed; (b) H. Kawai, T. Takeda, K. Fujiwara and T. Suzuki, Tetrahedron Lett., 2004, 45, 8289–8293 CrossRef CAS PubMed.
- T. Suzuki, S. Iwashita, T. Yoshino, H. Kawai, K. Fujiwara, M. Ohkita, T. Tsuji, K. Ono and M. Takenaka, Tetrahedron Lett., 2006, 47, 467–471 CrossRef CAS PubMed.
- A. Ishii, C. Tsuchiya, T. Shimada, K. Furusawa, T. Omata and J. Nakayama, J. Org. Chem., 2000, 65, 1799–1806 CrossRef CAS PubMed.
- (a) G. Dyker, K. Merz, I. M. Oppel and E. Muth, Synlett, 2007, 897–900 CrossRef CAS PubMed; (b) see also H. Kurata, Y. Takehara, S. Kim, K. Sakai, K. Matsumoto, T. Kawase and M. Oda, Synlett, 2007, 2053–2056 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data, details of X-ray single-crystal structure analyses, ORTEP drawings of 1–6, and Cartesian coordinates for the optimized structures of 1–6, acenaphthylene, acenaphthylene-5,6-diyldi(9-acridine) and acefluoranthylene. CCDC 975883–975890. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc49573f |
| This journal is © The Royal Society of Chemistry 2014 |