 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Versatile ruthenium(II)-catalyzed C–H cyanations of benzamides†
Weiping
Liu
and
Lutz
Ackermann
*
Institut fuer Organische und Biomolekulare Chemie, Georg-August-Universitaet, Tammannstrasse 2, 37077 Goettingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
First published on 6th January 2014
Abstract
Direct cyanations of arenes and heteroarenes bearing only weakly coordinating amides were accomplished using a robust ruthenium(II) catalyst. The user-friendly C(sp2)–H activation occurred with the assistance of carboxylate with high site-selectivity, excellent functional group tolerance and ample scope.
Aromatic nitriles constitute key structural motifs of important pharmaceuticals, dyes, agrochemicals and natural products.1–3 The cyano group serves as a versatile functional group that can easily be transformed into amines, ketones or aldehydes, among others.4 The syntheses of aryl nitriles continue to rely on classical approaches, such as the Sandmeyer5 or the Rosenmund–von Braun reaction,6,7 which have severe limitations, including the use of stoichiometric or even super-stoichiometric amounts of metal cyanides as well as harsh reaction conditions. An alternative approach is represented by transition metal-catalyzed cyanations of aryl halides or boronic acids under milder reaction conditions.8 These catalyzed coupling reactions thereby also display a considerably improved functional group tolerance. However, this strategy exploits prefunctionalized substrates, the preparation of which requires numerous functional group interconversions and generates undesired waste.
The catalytic activation of otherwise inert C(sp2)–H bonds as latent functional groups has in recent years emerged as an increasingly viable tool for improving the atom- and step-economy of organic synthesis.9 Thus, Yu developed direct cyanations of 2-arylpyridines.10,11 The research groups of Jiao12 and Chang13 reported on oxidative cyanations of electron-rich substrates with nonmetallic cyano-group sources in the presence of stoichiometric sacrificial oxidants,14,15 while Hartwig disclosed a two-step iridium-catalyzed borylation/copper-mediated cyanation protocol.16 Very recently, rhodium-catalyzed C–H cyanations of arenes were accomplished, albeit exploiting rather strongly coordinating directing groups, such as oxazolines, oximes, pyrazoles or pyridines.17–19 Herein, we wish to disclose an alternative strategy, which involves the use of relatively inexpensive20 ruthenium(II) catalysts21 for the first time for C(sp2)–H cyanations. Importantly, the versatile ruthenium catalyst enabled expedient C–H cyanations with easily accessible N-cyano-N-phenyl-p-toluenesulfonamide (NCTS, 2) as the most user-friendly cyanation reagent for the functionalization of only weakly coordinating22 aromatic and heteroaromatic amides – key structural scaffolds in organic synthesis and medicinal chemistry.
We initiated our studies by probing the effect of different additives and solvents on the envisioned C–H bond cyanation using synthetically useful benzamide 1a (Table 1).
|
|
|||
|---|---|---|---|
| Entry | Additive | Solvent | Yield (%) |
| a Reaction conditions: 1a (0.5 mmol), 2 (1.0 mmol), [RuCl2(p-cymene)]2 (5.0 mol%), AgSbF6 (20 mol%), additive (20 mol%), solvent (2.0 mL), 120 °C, 18 h; GC-conversion, isolated yields are given in parentheses. b AgOAc (30 mol%). c Without AgSbF6. d AgSbF6 (30 mol%). e Without [RuCl2(p-cymene)]2. f 24 h. | |||
| 1 | AgOAc | DCE | 77 (70) |
| 2 | Cu(OAc)2 | DCE | 30 |
| 3b | AgOAc | DCE | 80 (67) |
| 4c | AgOAc | DCE | <5 |
| 5d | AgOAc | DCE | 67 (54) |
| 6 | AgOAc | 1,4-Dioxane | 38 |
| 7 | AgOAc | o-Xylene | 21 |
| 8 | AgOAc | Toluene | 23 |
| 9 | AgOAc | DMF | <2 |
| 10e | AgOAc | DCE | <2 |
| 11 | KOAc | DCE | <5 |
| 12 | CsOAc | DCE | 17 |
| 13 | NaOAc | DCE | 90 (80) |
| 14 | NaOAc | DCE | 95 (84) |
Thus, [RuCl2(p-cymene)]2 enabled the desired direct cyanation when using both AgSbF6 and AgOAc as additives in DCE as the solvent (entries 1–5). DCE proved to be the solvent of choice (entries 1 and 6–9), while a test reaction illustrated that the C–H bond activation did not occur in the absence of the ruthenium catalyst (entry 10). Among a set of representative metal acetate additives,23,24 NaOAc furnished the highest yields of the product 3a (entries 11–14).
With the optimized catalytic system in hand, we explored the effect exerted by the N-substituent at the amide moiety (Scheme 1). A variety of tertiary amides 1a–1f proved to be suitable substrates with optimal results being accomplished with the sterically hindered substrate 1a.25 A comparable catalytic efficacy was obtained when performing the C–H bond cyanation on a larger 5 mmol scale.
 | ||
| Scheme 1 Effect of N-substituents on C–H cyanations. | ||
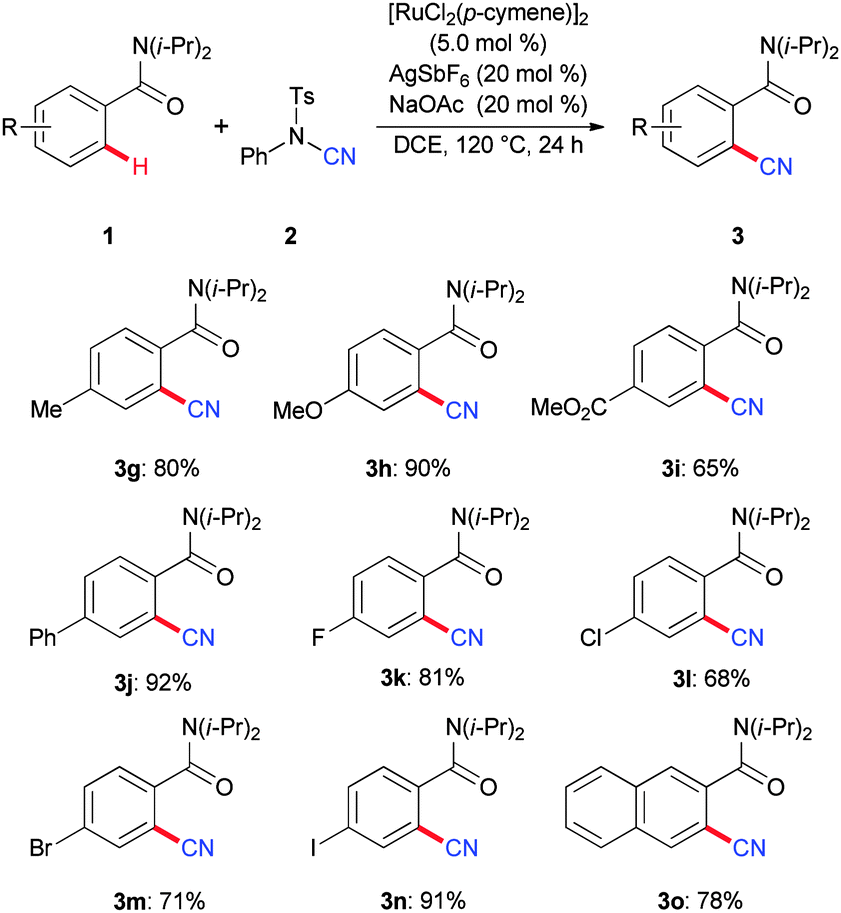
Subsequently, we tested the scope of the catalytic system for the cyanation of aromatic amides 1 (Scheme 2). The robust ruthenium(II) complex proved to be highly chemo-selective. Thus, the catalyst tolerated a set of valuable electrophilic functional groups, such as ester, fluoro, chloro, bromo and even iodo groups, which should be invaluable for the further diversification of the thus obtained products 3.
 | ||
| Scheme 2 Scope of ruthenium-catalyzed C–H cyanation with amides 1. | ||
The user-friendly ruthenium(II) catalyst was not limited to the functionalization of arenes. Indeed, challenging heteroaromatic substrates 1p–1u were efficiently directly functionalized both at positions C–2 (Scheme 3a) as well as C–3 (Scheme 3b). Thereby, cyanated thiophenes, furanes, benzothiophenes, benzofuranes and indoles were obtained in a chemo- and site-selective fashion.
 | ||
| Scheme 3 C–H cyanation with heteroaromatic amides 1. | ||
The well-defined ruthenium(II) biscarboxylate complex 426 displayed a catalytic activity comparable to the one observed when using the in situ generated system, notably even in the absence of NaOAc (Scheme 4).
 | ||
| Scheme 4 Well-defined complex 4 as the catalyst. | ||
In consideration of the unique chemo-selectivity and outstanding efficacy of our ruthenium catalyst, we became interested in delineating its mode of action. To this end, intramolecular competition experiments with meta-substituted substrates 1v–1x revealed a considerable secondary directing group effect27 exerted by a methoxy substituent and, even more pronounced, by a fluoro group present in benzamides 1w and 1x, respectively (Scheme 5).
 | ||
| Scheme 5 Intramolecular competition experiments with meta-substituted arenes 1. | ||
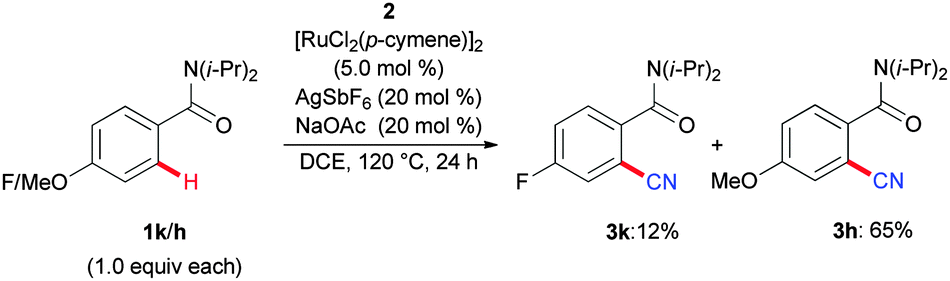
Intermolecular competition experiments between differently decorated amides indicated electron-rich arenes to be preferentially converted (Scheme 6), which can be rationalized in terms of an electrophilic-type activation mode of the cationic ruthenium species.
 | ||
| Scheme 6 Intermolecular competition experiments with arenes 1. | ||
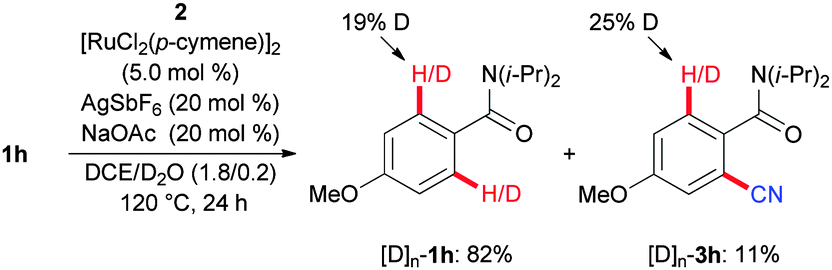
In support of this hypothesis, we observed a rather low kinetic isotope effect (KIE) of kH/kD ≈ 1.2 (see the ESI†). Moreover, C–H bond activation conducted in the presence of D2O as the cosolvent revealed a reversible H/D-exchange reaction, as was observed for the reisolated starting material [D]n-1h as well as the product [D]n-3h (Scheme 7).
 | ||
| Scheme 7 H/D exchange reaction. | ||
Based on these mechanistic studies we propose a plausible catalytic cycle to initiate a reversible C–H bond metalation on amides 128 to furnish cationic complex 6 (Scheme 8). Thereafter, coordination and insertion of NCTS (2) give rise to key intermediate 7. Subsequently, β-elimination provides the desired product 3, while proto-demetalation regenerates the cationic ruthenium(II) carboxylate catalyst 5.
 | ||
| Scheme 8 Plausible catalytic cycle. | ||
In summary, we have reported on the use of relatively inexpensive ruthenium-catalyzed C(sp2)–H bond cyanation on arenes for the first time. The robust direct cyanation29,30 occurred site-selectively on synthetically useful aromatic and heteroaromatic amides with ample scope. Experimental mechanistic studies provided strong support for a reversible C–H metalation mechanism by a cationic ruthenium(II) complex.
Support from the European Research Council under the European Community's Seventh Framework Program (FP7 2007–2013)/ERC Grant agreement no. 307535 and the Chinese Scholarship Council (fellowship to W.L.) is gratefully acknowledged.
Notes and references
- P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049 RSC.
- C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027 CrossRef CAS.
- M. Sundermeier, A. Zapf and M. Beller, Eur. J. Inorg. Chem., 2003, 3513 CrossRef CAS.
- R. C. Larock, Comprehensive Organic Transformations, Wiley-VCH, Weinheim, 1999 Search PubMed.
- T. Sandmeyer, Ber. Dtsch. Chem. Ges., 1884, 17, 1633 CrossRef.
- K. W. Rosenmund and E. Struck, Ber. Dtsch. Chem. Ges., 1919, 2, 1749 Search PubMed.
- J. von Braun and G. Manz, Justus Liebigs Ann. Chem., 1931, 488, 111 CrossRef.
- For selected examples, see: (a) T. D. Senecal, W. Shu and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 10035 CrossRef CAS PubMed; (b) P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 519 CrossRef CAS PubMed; (c) P. Anbarasan, H. Neumann and M. Beller, Chem.–Eur. J., 2011, 17, 4217 CrossRef CAS PubMed; (d) O. Grossman and D. Gelman, Org. Lett., 2006, 8, 1189 CrossRef CAS PubMed; (e) J. Zanon, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 2890 CrossRef CAS PubMed and references cited therein.
- Recent reviews on C–H bond functionalization: (a) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927 CrossRef CAS PubMed; (b) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (c) X. Shang and Z.-Q. Liu, Chem. Soc. Rev., 2013, 42, 3253 RSC; (d) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177 CrossRef CAS PubMed; (e) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (f) L. Ackermann, Chem. Commun., 2010, 46, 4866 RSC; (g) M. Livendahl and A. M. Echavarren, Isr. J. Chem., 2010, 50, 630 CrossRef CAS; (h) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS PubMed and cited references.
- X. Chen, X. S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed.
- For pioneering functionalizations of tertiary amines, see: (a) S.-I. Murahashi, T. Nakae, H. Terai and N. Komiya, J. Am. Chem. Soc., 2008, 130, 11005 CrossRef CAS PubMed; (b) S.-I. Murahashi, N. Komiya and H. Terai, Angew. Chem., Int. Ed., 2005, 44, 6931 CrossRef CAS PubMed; (c) S.-I. Murahashi, N. Komiya, H. Terai and T. Nakae, J. Am. Chem. Soc., 2003, 125, 15312 CrossRef CAS PubMed and cited references.
- S. Ding and N. Jiao, J. Am. Chem. Soc., 2011, 133, 12374 CrossRef CAS PubMed.
- J. Kim, J. Choi, K. Shin and S. Chang, J. Am. Chem. Soc., 2012, 134, 2528 CrossRef CAS PubMed.
- S. Ding and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 9226 CrossRef CAS PubMed.
- J. Kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948 CrossRef CAS PubMed.
- C. W. Liskey, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 11389 CrossRef CAS PubMed.
- T.-J. Gong, B. Xiao, W.-M. Cheng, W. Su, J. Xu, Z.-J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 10630 CrossRef CAS PubMed.
- M. Chaitanya, D. Yadagiri and P. Anbarasan, Org. Lett., 2013, 15, 4960 CrossRef CAS PubMed.
- See also: (a) Y. Yang, Y. Zhang and J. Wang, Org. Lett., 2011, 13, 5608 CrossRef CAS PubMed; (b) H.-Q. Do and O. Daugulis, Org. Lett., 2010, 12, 2517 CrossRef CAS PubMed.
- The prices of platinum, rhodium, gold, iridium, palladium and ruthenium were 1482, 1015, 1363, 800, 701 and 80 US$ per troy oz, respectively, as of September 2013. See: 〈http://taxfreegold.co.uk/preciousmetalpricesusdollars.html〉.
- For recent reviews on ruthenium-catalyzed C–H functionalization, see: (a) V. S. Thirunavukkarasu, S. I. Kozhushkov and L. Ackermann, Chem. Commun., 2014, 50, 29 RSC; (b) L. Ackermann, Acc. Chem. Res., 2013, 46 DOI:10.1021/ar3002798; (c) B. Li and P. H. Dixneuf, Chem. Soc. Rev., 2013, 42, 5744 RSC; (d) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886 RSC; (e) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (f) L. Ackermann, Pure Appl. Chem., 2010, 82, 1403 CrossRef CAS; (g) F. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826 CrossRef CAS PubMed and cited references.
- K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed.
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef.
- The mass balance of the cyanation reactions with substrates 1b–1f is accounted for by the unreacted starting materials.
- L. Ackermann, P. Novák, R. Vicente and N. Hofmann, Angew. Chem., Int. Ed., 2009, 48, 6045 CrossRef CAS PubMed.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749 CrossRef CAS PubMed.
- Preliminary experiments with anilides or aryl carbamates as the substrates provided thus far less satisfactory results.
- For recent examples of ruthenium(II)-catalyzed C–H oxygenations, see: (a) W. Liu and L. Ackermann, Org. Lett., 2013, 15, 3484 CrossRef CAS PubMed; (b) F. Yang and L. Ackermann, Org. Lett., 2013, 15, 718 CrossRef CAS PubMed; (c) V. S. Thirunavukkarasu and L. Ackermann, Org. Lett., 2012, 14, 6206 CrossRef CAS PubMed; (d) V. S. Thirunavukkarasu, J. Hubrich and L. Ackermann, Org. Lett., 2012, 14, 4210 CrossRef CAS PubMed; (e) Y. Yang, Y. Lin and Y. Rao, Org. Lett., 2012, 14, 2874 CrossRef CAS PubMed.
- A recent ruthenium(II)-catalyzed C–H halogenation: L. Wang and L. Ackermann, Chem. Commun., 2014, 50, 1083 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, and 1H and 13C NMR spectra of products. See DOI: 10.1039/c3cc49502g |
| This journal is © The Royal Society of Chemistry 2014 |