 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective, intermolecular [2+2] photocycloaddition reactions of 3-acetoxyquinolone: total synthesis of (−)-pinolinone†
Florian
Mayr
,
Christian
Wiegand
and
Thorsten
Bach
*
Lehrstuhl für Organische Chemie I, Technische Universität München, 85747 Garching, Germany. E-mail: thorsten.bach@ch.tum.de; Fax: +49 89 28913315; Tel: +49 89 28913330
First published on 18th February 2014
Abstract
The natural product (−)-pinolinone was synthesised via a concise route (six steps, 17% overall yield) from 3-acetoxyquinolone, employing an enantioselective intermolecular [2+2] photocycloaddition as the key step.
(−)-Pinolinone was described in 2000 as a novel, naturally occurring 3,4-dihydroquinolin-2(1H)-one, which was isolated by extracting dried roots of Boronia pinnata Sm. (Rutaceae) with acetone at ambient temperature.1 It belongs to the class of 3-prenylated quinolin-2(1H)-ones, some representatives of which had been previously reported,2 but only one as a constituent of the plant genus Boronia.3 3,4-Dihydroxylation is observed in many naturally occurring 3,4-dihydroquinolin-2(1H)-ones derived from anthranilic acid. However, most of these compounds bear an additional 4-aryl substituent derived from an aromatic amino acid.4
Despite the fact that little information was available on the biological activity of (−)-pinolinone, we became interested in its total synthesis with the goal of proving its constitution and its absolute configuration. Indeed, although the relative configuration of the natural product had been established, there was no information about its absolute configuration and both structures 1 (Scheme 1) and ent-1 were conceivable.
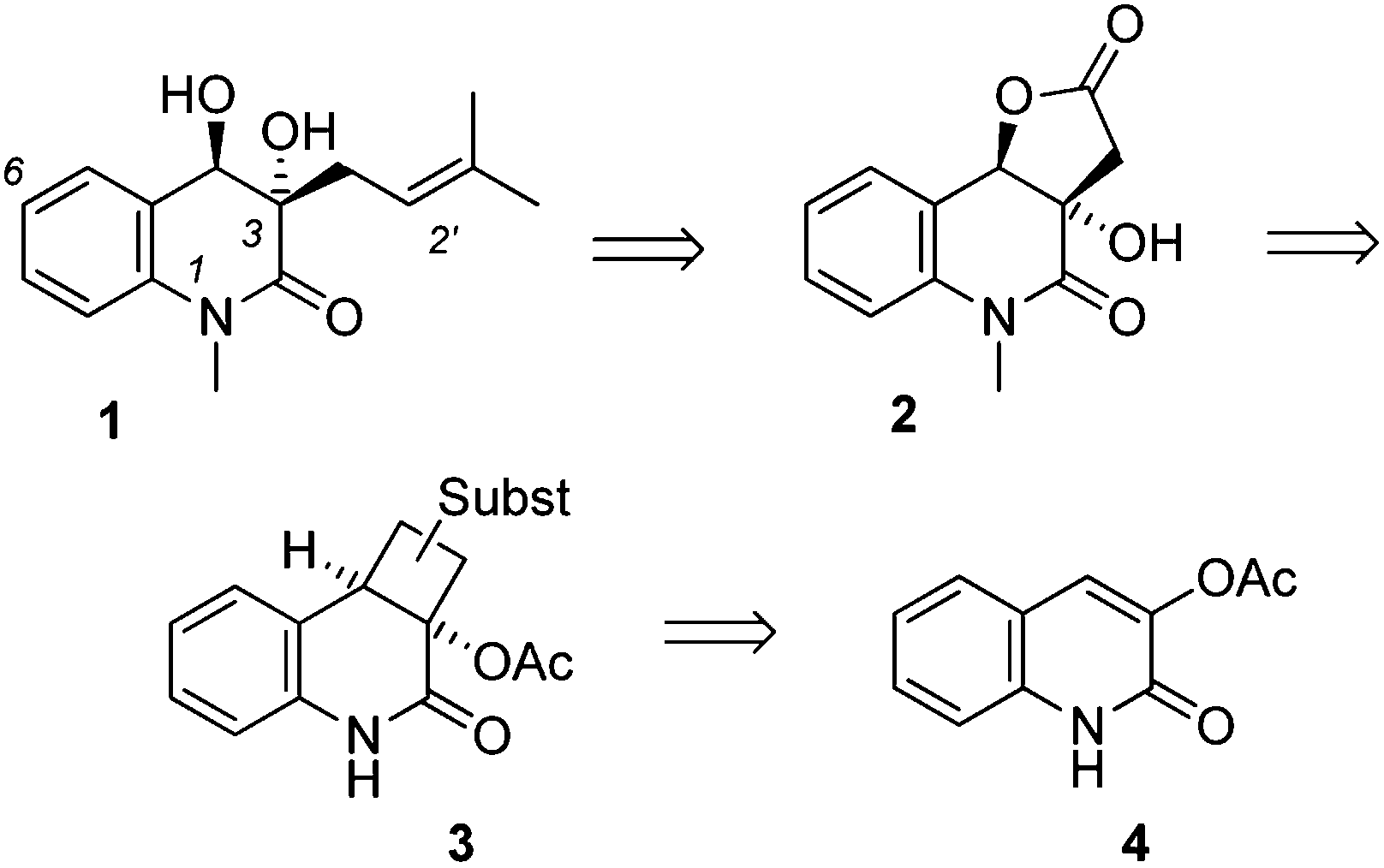 | ||
| Scheme 1 Relative configuration of pinolinone (1) (ref. 1) and retrosynthetic disconnection leading via intermediates 2 and 3 (Subst = substituents) to the title compound, 3-acetoxyquinolone (4). | ||
Based on the successful implementation of an enantioselective,5 intermolecular [2+2] photocycloaddition approach6 in a previous total synthesis7 of a 3,4-dihydroquinolin-2(1H)-one, (+)-meloscine,8 we considered the addition of an appropriate olefin to 3-acetoxyquinolone (4) to be an efficient way to set up the two stereogenic centers of the natural product. Intermediate 3 was to be converted into γ-lactone 2, from which the target compound would be accessible by straightforward transformations. In this communication we disclose the successful execution of this photocycloaddition strategy, which led to the first synthesis of (−)-pinolinone and allowed for the assignment of its absolute configuration.
Previous work by Kobayashi et al. has revealed that 3-acetoxyquinolone (4) is a competent enone substrate in intermolecular [2+2] photocycloaddition reactions.9 Successful reactions with cyclic and acyclic alkenes as well as with an enol ether (2-methoxypropene) were reported. These results are in line with the observation that electron-rich alkenes are good reaction partners in intermolecular quinolone [2+2] photocycloaddition reactions.10 Our plan was to find an appropriate ketene acetal, which would react with sufficient regioselectivity to provide, after hydrolysis a stereo- and regiochemically homogenous cyclobutanone, the Baeyer–Villiger oxidation11 of which would lead to the desired γ-lactone. A second goal of the preliminary work was to probe the suitability of the title compound for enantioselective [2+2] photocycloaddition reactions in the presence of chiral template 6 (ref. 12) (Table 1). Among the ketene acetals, which were tested in the racemic reaction (λ = 366 nm, c = 5 mM in acetonitrile) 1,1-diethoxyethene (5e) and silyl enol ether 5f turned out to be best suited. Other olefins that also reacted well and which had not been previously studied included vinyl acetate (5a), 1,1-dichloroethene (5b), 2-ethyl-1-butene (5c) and vinylene carbonate. Yields varied between 62% and 99% (see the ESI† for additional information). Almost any of the mentioned olefins – apart from vinylidene carbonate – and also the previously studied 2,3-dimethyl-2-butene (5d) reacted equally well in the templated reaction at −70 °C in toluene. The enantioselectivities were somewhat variable but – given the fact that 3-alkenyloxy-substituted quinolones had shown relatively low enantioselectivities in template mediated intramolecular [2+2] photocycloaddition reactions13 – it was gratifying to note that in particular the ketene acetals 5e and 5f reacted smoothly with excellent enantioselectivites. Product 3e was obtained in 67% yield and with 95% ee and product 3f was obtained in 97% yield with 88% ee for the major diastereoisomer and with 95% ee for the minor diastereoisomer (d.r. = 71/29).
|
|
|---|
| a All photochemical reactions were conducted using a RPR-100 reactor with 16 fluorescent lamps (Duran apparatus; for the emission spectrum, see ref. 13) as the irradiation source in deaerated toluene (c = 5 mM) as the solvent. The irradiation time was three hours. Yields refer to isolated products. b Diastereomeric ratio for the two diastereoisomers. For structure assignments, see the narrative and ESI. c Enantiomeric excess of major and minor diastereoisomers, respectively. |

|
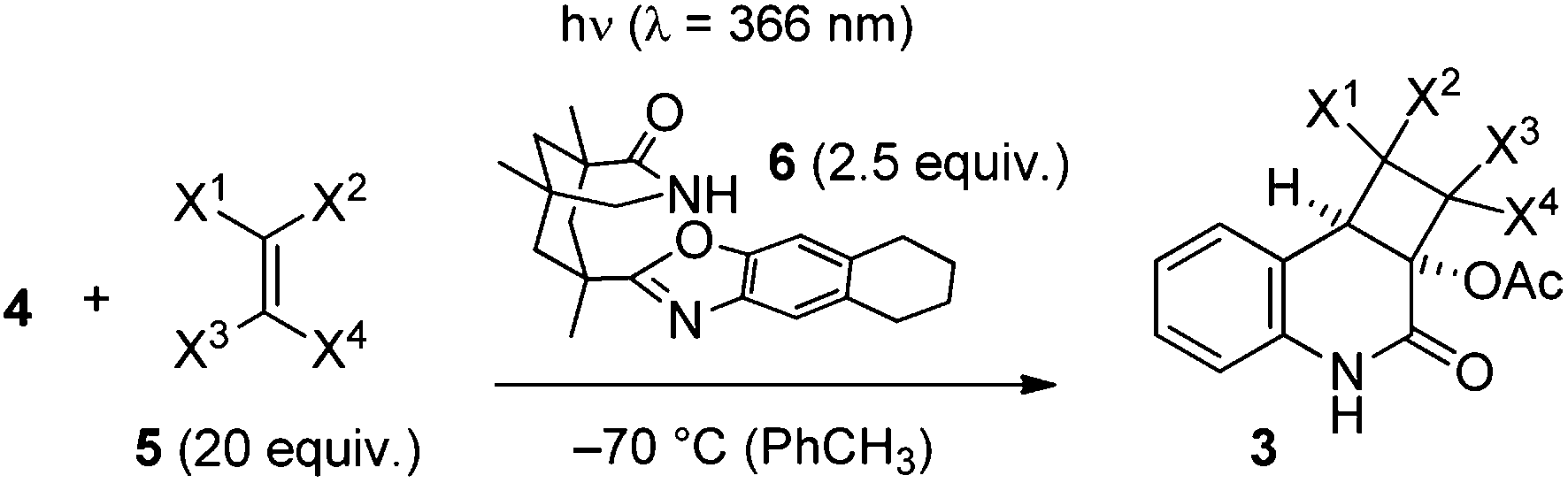
The regioselectivity of the photocycloaddition was high and products 3a, 3b, 3e and 3f were potentially suited for conversion into the desired cyclobutanone. The high enantioselectivities achieved for 3e and 3f and the fact that there would be no change of the oxidation state required for 3e and 3f (as opposed to 3a) led us to study the latter two products for conversion into a cyclobutanone. Preliminary experiments were performed with racemic materials. The N-methylation of 3,4-dihydroquinolin-2(1H)-one rac-3e was facile, employing NaH and MeI in THF (84% yield)14 but conditions for the smooth hydrolysis of the O,O-acetal could not be found. There was no reaction upon treatment with aqueous HCl or with para-toluenesulfonic acid in THF while an attempted reaction with trifluoroacetic acid led to complete decomposition. Similar observations were made with acetal rac-3f. Neither the compound itself nor its N-methylated derivative could be converted into the desired cyclobutanone (Scheme 2). A major issue with these transformations is the facile retro-aldol cleavage, which can occur in compounds such as rac-7 under basic conditions and which leads to a cyclobutane ring opening. It was suspected that the presence of fluoride ions required for desilylation induces a deacetylation of the tertiary alcohol, which in turn leads to decomposition. Variation of acidic conditions for tert-butylsilyldimethylsilyl (TBS) removal led to the discovery that the protecting group removal can be combined with the desired oxidative Baeyer–Villiger type rearrangement employing meta-chloroperbenzoic acid (mCPBA) as the oxidant in the presence of boron trifluoride as the Lewis acid.15 Although lactone rac-8 was isolated in low yields this discovery opened a viable route for completion of the total synthesis.
 | ||
| Scheme 2 Unsuccessful attempts to convert compound rac-3f into cyclobutanone rac-7 and successful oxidation to lactol rac-8. | ||
After N-methylation of enantiomerically enriched compound 3f16 the resulting product 9 underwent the desired oxidative rearrangement in 69% yield. However, it was not possible to perform the required simultaneous reduction of the lactone and the acetate to the desired product 11 in good yields. The sequence was therefore performed stepwise, with the deacetylation preceding the oxidation. Tertiary alcohol 10 was obtained from acetate 9 upon treatment with KCN in ethanol17 and was then oxidized with mCPBA to γ-lactone 2 (Scheme 3). At this stage, the compound (87% ee) was purified by semipreparative chiral HPLC to obtain enantiopure material (≥99% ee).
 | ||
| Scheme 3 Synthetic sequence to (−)-pinolinone (1) from enantiomerically enriched [2+2] photocycloaddition product 3f. | ||
In the subsequent reduction of lactone 2 to lactol 11, it was observed that the success of the reaction depends strongly on the solvent and the number of equivalents of diisobutylaluminium hydride (DIBAL-H).18 Dichloromethane turned out to be superior to hexane, toluene or ether as the solvent. For complete conversion three equivalents of the reducing agent were required. Lactol 11, which was obtained under optimized conditions in 61% yield, should be in equilibrium with the respective γ-hydroxyaldehyde and it was therefore assumed that a Wittig reaction could be successfully executed. Despite the extensive literature precedence that exists for this transformation,19 there is no reaction, in which a γ-lactol was converted to a triply substituted olefin with two methyl groups at the terminal position. A further complication in the projected transformation was the acidity of the tertiary alcohol. Initial attempts to achieve the reaction by addition of the ylide to the lactol remained unsuccessful but it was found that the reaction could be performed at 0 °C by adding the lactol to an excess of the ylide in THF as the solvent. Under these conditions, a yield of 55% could be achieved for the olefination reaction and product 1 was shown to be identical to pinolinone based on its analytical data. The compound was found to be levorotatory, which – based on the known enantioselectivity exerted by template 6 (ref. 6 and 12) – proved the absolute configuration to be (3S,4R). Despite the fact that the enantiopurity of final product 1 was confirmed by chiral HPLC, the specific rotation was lower than the reported value.20
In summary, the first total synthesis of (−)-pinolinone (1) has been accomplished via an enantioselective intermolecular [2+2] photocycloaddition of 3-acetoxyquinolone (4) and an appropriate silyl ketene acetal. Further key steps included a selective ring expansion of the cyclobutane ring to a γ-lactone and the subsequent conversion of the latter compound by a reduction/olefination sequence.
This project was supported by the Deutsche Forschungsgemeinschaft (Ba 1372-10, GRK 1626), by the TUM Graduate School, and by the Fonds der Chemischen Industrie. Olaf Ackermann and Marcus Wegmann are acknowledged for help with the HPLC analyses.
Notes and references
- C. Ito, M. Itoigawa, T. Otsuka, H. Tokuda, H. Nishino and H. Furukawa, J. Nat. Prod., 2000, 63, 1344–1348 CrossRef CAS PubMed.
- D. Lavie, N. Danieli, R. Weitman and E. Glotter, Tetrahedron, 1968, 24, 3011–3018 CrossRef CAS.
- M. Ahsan, A. I. Gray and P. G. Waterman, J. Nat. Prod., 1994, 57, 670–672 CrossRef CAS.
- (a) Y. Kimura, M. Kusano, H. Koshino, J. Uzawa, S. Fujioka and K. Tani, Tetrahedron Lett., 1996, 37, 4961–4964 CrossRef CAS; (b) R. Uchida, R. Imasato, K. Shiomi, H. Tomoda and S. Ōmura, Org. Lett., 2005, 7, 5701–5704 CrossRef CAS PubMed; (c) K. Scherlach and C. Hertweck, Org. Biomol. Chem., 2006, 4, 3517–3520 RSC; (d) R. Uchida, R. Imasato, H. Tomoda and S. Ōmura, J. Antibiot., 2006, 59, 652–658 CrossRef CAS PubMed; (e) S. A. Neff, S. U. Lee, Y. Asami, J. S. Ahn, H. Oh, J. Baltrusaitis, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 2012, 75, 464–472 CrossRef CAS PubMed.
- For recent reviews on enantioselective photochemical reactions, see: (a) C. Yang and Y. Inoue, Chem. Soc. Rev., 2014, 43 10.1039/c3cs60339c; (b) C. Müller and T. Bach, Aust. J. Chem., 2008, 61, 557–564 CrossRef.
- (a) T. Bach and H. Bergmann, J. Am. Chem. Soc., 2000, 122, 11525–11526 CrossRef CAS; (b) T. Bach, H. Bergmann, B. Grosch and K. Harms, J. Am. Chem. Soc., 2002, 124, 7982–7990 CrossRef CAS PubMed.
- For reviews on the use of [2+2] photocycloaddition reactions in synthesis, see: (a) J. Iriondo-Alberdi and M. F. Greaney, Eur. J. Org. Chem., 2007, 4801–4815 CrossRef CAS; (b) N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed; (c) T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed.
- (a) P. Selig and T. Bach, Angew. Chem., Int. Ed., 2008, 47, 5082–5084 CrossRef CAS PubMed; (b) P. Selig, E. Herdtweck and T. Bach, Chem.–Eur. J., 2009, 15, 3509–3525 CrossRef CAS PubMed.
- (a) H. Suginome, K. Kobayashi, M. Itoh and S. Seko, J. Org. Chem., 1990, 55, 4933–4943 CrossRef CAS; (b) K. Kobayashi, M. Suzuki and H. Suginome, J. Org. Chem., 1992, 57, 599–606 CrossRef CAS.
- (a) B. Loev, M. M. Goodman and K. M. Snader, Tetrahedron Lett., 1968, 9, 5401–5404 CrossRef; (b) T. S. Cantrell, J. Org. Chem., 1974, 39, 3063–3070 CrossRef CAS; (c) C. Kaneko, T. Naito and M. Somei, J. Chem. Soc., Chem. Commun., 1979, 804–805 RSC; (d) T. Naito and C. Kaneko, Chem. Pharm. Bull., 1980, 28, 3150–3152 CrossRef CAS; (e) T. Naito, Y. Momose and C. Kaneko, Chem. Pharm. Bull., 1982, 30, 1531–1534 CrossRef CAS; (f) C. Kaneko, N. Shimomura, Y. Momose and T. Naito, Chem. Lett., 1983, 1239–1242 CrossRef CAS; (g) C. Kaneko, T. Suzuki, M. Sato and T. Naito, Chem. Pharm. Bull., 1987, 35, 112–123 CrossRef CAS; (h) E. Sato, Y. Ikeda and Y. Kanaoka, Liebigs Ann. Chem., 1989, 781–788 CrossRef CAS; (i) F. Yagishita, T. Mino, T. Fujita and M. Sakamoto, Org. Lett., 2012, 14, 2638–2641 CrossRef CAS PubMed.
- Reviews: (a) C. H. Hassall, Org. React., 1957, 9, 73–106 Search PubMed; (b) G. C. Krow, Org. React., 1993, 43, 251–798 CAS; (c) M. Renz and B. Meunier, Eur. J. Org. Chem., 1999, 737–750 CrossRef CAS; (d) G.-J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105–4123 CrossRef PubMed.
- (a) T. Bach, H. Bergmann and K. Harms, Angew. Chem., Int. Ed., 2000, 39, 2302–2304 CrossRef CAS; (b) T. Bach, H. Bergmann, B. Grosch, K. Harms and E. Herdtweck, Synthesis, 2001, 1395–1405 CrossRef CAS PubMed.
- M. M. Maturi, M. Wenninger, R. Alonso, A. Bauer, A. Pöthig, E. Riedle and T. Bach, Chem.–Eur. J., 2013, 19, 7461–7472 CrossRef CAS PubMed.
- (a) M. Uchida, F. Tabusa, M. Komatsu, S. Morita, T. Kanbe and K. Nakagawa, Chem. Pharm. Bull., 1985, 33, 3775–3786 CrossRef CAS; (b) F. Voss, F. Vogt, E. Herdtweck and T. Bach, Synthesis, 2011, 961–971 CAS.
- For the use of a Lewis acid (SnCl4) in an enantioselective Baeyer–Villiger oxidation approach employing a chiral ketal, see: T. Sugimura, Y. Fujiwara and A. Tai, Tetrahedron Lett., 1997, 38, 6019–6022 CrossRef CAS.
- The starting material for the total synthesis was generated by enantioselective [2+2] photocycloaddition reactions, which were not consistently performed at −70 °C but – due to insufficient cooling – also at slightly elevated temperatures. As known from previous work6,12 and as detailed in the ESI† the enantioselectivity decreases with increasing irradiation temperature.
- (a) G. J. F. Chittenden, Carbohydr. Res., 1991, 222, 283–287 CrossRef CAS; (b) C. C. Joseph, H. Regeling, B. Zwanenburg and G. J. F. Chittenden, Tetrahedron, 2002, 58, 6907–6911 CrossRef CAS; (c) L. Kværnø, M. Werder, H. Hauser and E. M. Carreira, J. Med. Chem., 2005, 48, 6035–6053 CrossRef PubMed; (d) S. Hanessian, L. Auzzas, G. Giannini, M. Marzi, W. Cabri, M. Barbarino, L. Vesci and C. Pisano, Bioorg. Med. Chem. Lett., 2007, 17, 6261–6265 CrossRef CAS PubMed.
- (a) M. Babjak, P. Kapitán and T. Gracza, Tetrahedron, 2005, 61, 2471–2479 CrossRef CAS PubMed; (b) M. Shoji, T. Uno, H. Kakeya, R. Onose, I. Shiina, H. Osada and Y. Hayashi, J. Org. Chem., 2005, 70, 9905–9915 CrossRef CAS PubMed; (c) N. Joubert, R. Pohl, B. Klepetářová and M. Hocek, J. Org. Chem., 2007, 72, 6797–6805 CrossRef CAS PubMed; (d) D. Enders, A. Hieronymi and G. Raabe, Synthesis, 2008, 1545–1558 CrossRef CAS PubMed.
- Examples: (a) P. A. Grieco and J. J. Reap, J. Org. Chem., 1973, 38, 3413–3415 CrossRef CAS; (b) J.-P. Deprés, A. E. Greene and P. Crabbé, Tetrahedron, 1981, 37, 621–628 CrossRef; (c) L. van Hijfte, R. D. Little, J. L. Petersen and K. D. Moeller, J. Org. Chem., 1987, 52, 4647–4661 CrossRef CAS; (d) G. V. M. Sharma and S. M. Rao, Tetrahedron Lett., 1992, 33, 2365–2368 CrossRef CAS; (e) Y. Kimura, M. Kusano, H. Koshino, J. Uzawa, S. Fujioka and K. Tani, Tetrahedron Lett., 1996, 37, 4961–4964 CrossRef CAS; (f) M. Ernst and G. Helmchen, Angew. Chem., Int. Ed., 2002, 41, 4054–4056 CrossRef CAS; (g) L. S. Hegedus, L. Geisler, A. G. Riches, S. S. Salman and G. Umbricht, J. Org. Chem., 2002, 67, 7649–7655 CrossRef CAS PubMed.
- The reported value for the specific rotation was [α]20D = − 9.4 (c = 0.128, CHCl3).1 For the synthetic material the specific rotation was determined as [α]20D = −6.3 (c = 0.16, CHCl3).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc49469a |
| This journal is © The Royal Society of Chemistry 2014 |