 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A sulfur mimic of 1,1-bis(diphenylphosphino)methane: a new ligand opens up†
Peter E.
Sues
,
Alan J.
Lough
and
Robert H.
Morris
*
Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: rmorris@chem.utoronto.ca
First published on 19th March 2014
Abstract
A simple method for synthesizing diphosphine monosulfide species was developed utilizing lithium sulfide and chlorophosphine starting materials. This afforded 1,1,2,2-tetraphenyldiphosphine monosulfide (1), as well as 1,1,2,2-tetracyclohexyldiphosphine monosulfide (2), which could be used as convenient ligand precursors. Upon addition of 1 or 2 to the ruthenium compound Ru(C5Me5)(cod)Cl, the diphosphine monosulfides rearranged to give bidentate bis(ditertiaryphosphino)thioether ligands in Ru(C5Me5)(PPh2SPPh2)Cl (3) and Ru(C5Me5)(PCy2SPCy2)Cl (4).
Bidentate phosphorus ligands have been widely employed by inorganic chemists in the synthesis of a large variety of metal complexes.1 A particularly noteworthy member of this class of ligands is 1,1-bis(diphenylphosphino)methane (dppm), which has been used, along with its alkyl and aryl substituted analogues, to generate monometallic2 and bimetallic (A-frame) compounds,3 as well as larger metal clusters.4 Moreover, many of these dppm-containing species were found to be active catalysts for hydroformylation and hydrocyanation reactions, as well as other chemical transformations.5–7
A related class of ligands is constituted by bis(ditertiaryphosphino)thioethers, which have a sulfur atom replacing the methylene backbone. Typically, synthetic strategies utilizing low temperatures and/or large fluorinated or aliphatic substituents on the phosphorus centres have been employed to stabilize the free thioether species, but despite these efforts, a diphosphine monosulfide byproduct is also commonly seen.8–42 In addition, although much has been done in synthesizing and isolating these types of molecules there are few examples of their metal complexes. To our knowledge only Burg et al. and Arnold et al. have reported nickel–25,41 and molybdenum–carbonyl22,43 complexes, respectively.
Although bis(ditertiaryphosphino)thioethers superficially resemble dppm, their stability and electronic properties are unlikely to be the same. P–S bonds are chemical analogues of P–O bonds and it is known that phosphines and phosphites have drastically different properties.44,45 Phosphites are less sigma donating and are more pi acidic, while phosphines have the opposite properties.44 Moreover, P–C bonds are much more stable than P–O bonds, which are sensitive to hydrolysis, alcoholysis, and alkoxide substitution reactions.45 As such, bis(ditertiaryphosphino)thioethers may have unique bonding properties that could be useful in tuning the electronic nature of a variety of transition metal catalysts.
In this paper we present a very simple method for synthesizing the previously reported diphosphine monosulfide species 1,1,2,2-tetraphenyldiphosphine monosulfide, 1, which was formerly synthesized using thiourea and chlorodiphenylphosphine.24,39 One equivalent of lithium sulfide was dissolved in acetonitrile, and two equivalents of chlorodiphenylphosphine were added, which afforded 1 as a white powder in high yields (87%, see Fig. 1). The 31P{1H} NMR spectrum of 1 was very diagnostic with two doublets at 42 and −16 ppm displaying an extremely large JPP coupling constant of 247 Hz (in agreement with literature values), indicating the presence of a P–P bond.24,39 The structure of 1 was also determined by single crystal X-ray diffraction (XRD, see Fig. S1, ESI†), which matched the results reported by Aluri et al. with a P–P bond length of 2.226(2) Å and a P–S bond length of 1.953(2) Å (see Table S1 (ESI†) for other notable bond lengths and angles).46
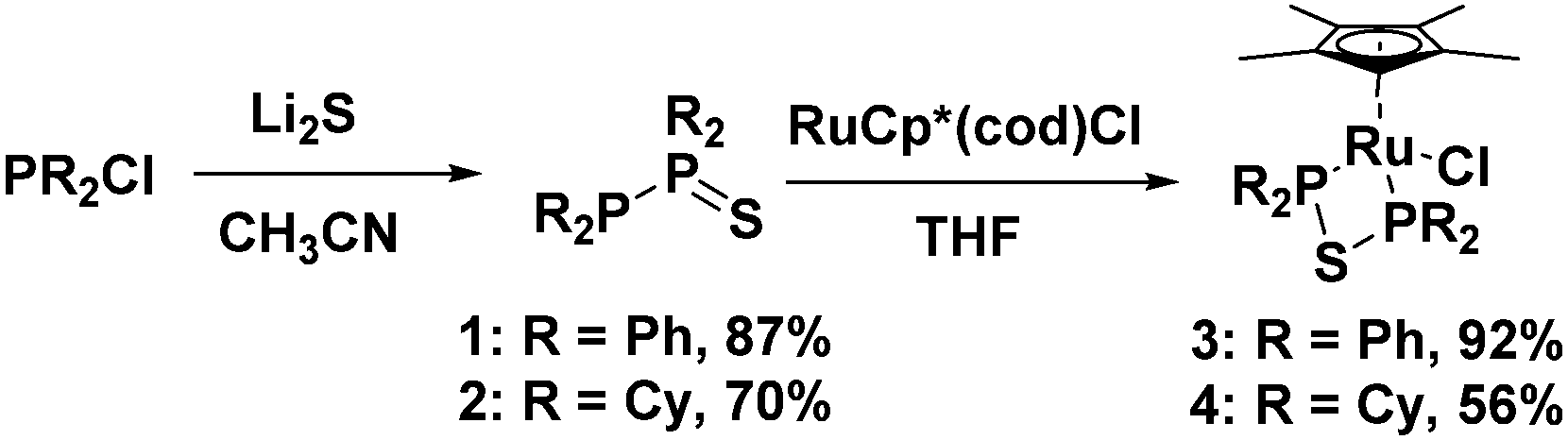 | ||
| Fig. 1 Synthesis of the monosulfides 1 and 2, as well as ruthenium complexes 3 and 4. | ||
Using the same synthetic methodology employed in the production of 1, an analogous alkyl substituted compound, 1,1,2,2-tetracyclohexyldiphosphine monosulfide, 2, was generated as a white powder in moderate yields (70%) utilizing chlorodicyclohexylphosphine as a starting material. The 31P{1H} NMR spectrum of 2 was very similar to that of 1 with two doublets at 59.1 and −14.0 ppm and a JPP coupling constant of 302 Hz. The XRD structure of the cyclohexyl-substituted analogue was also similar to that of the phenyl-substituted compound with a P–P bond length of 2.225(4) Å and a P–S bond length of 1.972(2) Å (see Fig. 2, and see Table S2 (ESI†) for other notable bond lengths and angles).
 | ||
| Fig. 2 ORTEP3 representation and atom numbering for 2 (thermal ellipsoids at 50% probability; all the hydrogens are omitted for clarity). | ||
Unexpectedly, given that a chromium complex bearing a 1,1,2,2-tetraphenyldiphosphine monosulfide ligand is known in the literature,47 diphosphine monosulfides 1 and 2 were found to be convenient precursors for metal complexes bearing bis(diphenylphosphino)thioether (dppte) and bis(dicyclohexylphosphino)thioether (dcpte) ligands, respectively. Treatment of RuCp*(cod)Cl (cod = 1,5-cyclooctadiene) with 1,1,2,2-tetraphenyldiphosphine monosulfide generated a neutral ruthenium complex RuCp*((PPh2)2S)Cl, 3, which could be isolated as a yellow powder in excellent yield (92%, see Fig. 1), while 1,1,2,2-tetracyclohexyldiphosphine monosulfide gave RuCp*((PCy2)2S)Cl, 4, as a yellow powder in 56% yield (see Fig. 1). The 31P{1H} NMR spectrum of 3 and 4 were very informative as only one peak could be seen at 39 ppm and 67 ppm, respectively, demonstrating that the inequivalent phosphorus moieties in the initial ligand precursors had become chemically equivalent. In addition, the ruthenium dppte and dcpte species were found to be tolerant of a wide variety of solvents, including acetonitrile, acetone, dichloromethane, methanol, ethanol, and isopropanol. The tolerance to alcohol solvents is of particular interest due to the sensitivity of other P–O and P–S bonds to alcoholysis and alkoxide substitution.45,48
A single crystal XRD study showed that 3 displays a piano-stool structure with Cp* occupying one half of the coordination sphere, while a chloride ligand along with dppte are bound at the other three coordination sites (see Fig. 3a). The bidentate ligand has a P(1)–Ru(1)–P(2) bite angle of 75.8(2)°, which is larger than analogous dppm structures, which have bite angles around 72°.2,43 The P(1)–S(1)–P(2) bond angle is quite acute, 82.1(2)°, which is more compressed than the P–S–P bond angle of 86.9(1)° found in a monometallic molybdenum carbonyl complex bearing a single dppte ligand (see Table S2 (ESI†) for other notable bond lengths and angles).43 Moreover, the P(1)–S(1)–P(2) bond angle is also significantly smaller than the P–C–P bond angle found in monometallic dppm structures (around 97°).2
 | ||
| Fig. 3 ORTEP3 representation and atom numbering for (a) 3 (thermal ellipsoids at 50% probability the solvent molecules and all the hydrogens are omitted for clarity); and (b) 4 (thermal ellipsoids at 50% probability all the hydrogens are omitted for clarity). | ||
Complex 4 was also characterized by XRD, and displays a similar coordination geometry as complex 3 (see Fig. 3b). The bidentate dcpte ligand has a P(1)–Ru(1)–P(2) bite angle of 74.71(6)° and a P(1)–S(1)–P(2) bond angle of 82.19(9)°, which are very similar to the metrical parameters seen for the dppte ligand in 3. The P(1)–Ru(1) and P(2)–Ru(1) bond lengths of 2.315(2) and 2.302(2) Å, respectively, however, are longer than the P(1)–Ru(1) and P(2)–Ru(1) bond lengths of 2.288(4) and 2.278(4) Å, respectively, seen for complex 3 (see Table S3 (ESI†) for other notable bond lengths and angles). This increase in bond length is most likely caused by the more sterically demanding cyclohexyl substituents, which are pushed away by the Cp* methyl groups.
Based on our coordination studies we propose that the ligand precursors 1 and 2 exist in equilibrium with their corresponding thioether constitutional isomers in solution (see Fig. 4). Initially, the diphosphinothioether likely forms from an intermediate monophosphine monosulfide species, but the diphosphine monosulfide form is significantly more stable. This drives the equilibrium far to the right, and therefore only 1 and 2 are seen.
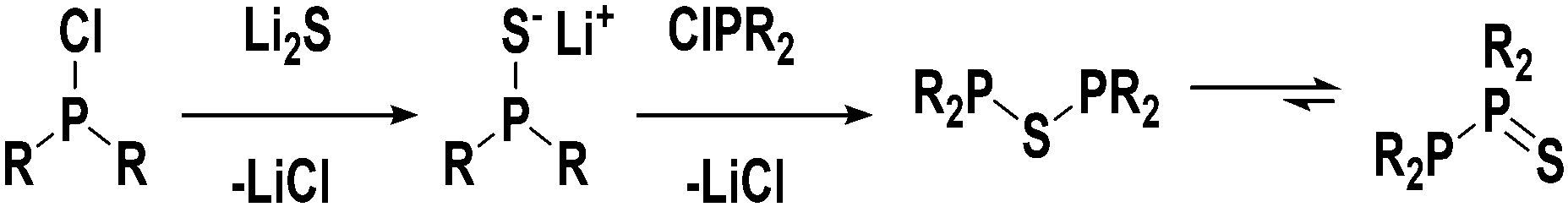 | ||
| Fig. 4 Proposed mechanism for the formation of 1 and 2. | ||
When the RuCp*(cod)Cl complex is introduced into the system, we believe that the diphosphine monosulfides coordinate to the metal first. There is precedent in the literature for this type of structure in the form of the chromium complex discussed previously.47 Once coordinated, the diphosphine monosulfide ligands can still interconvert into their thioether isomers and “open up”. When this happens, however, the metal centre traps the thioether as a bidentate ligand, and the equilibrium (Fig. 5) is forced to the right. As such, this reaction is likely to be very general and a wide variety of metal precursors should be suitable for this ligand architecture. The most crucial requirement, though, is that the metal has at least one vacant site to facilitate initial coordination of the diphosphine monosulfide precursor, and then the ability to make another site available to trap the thioether species.
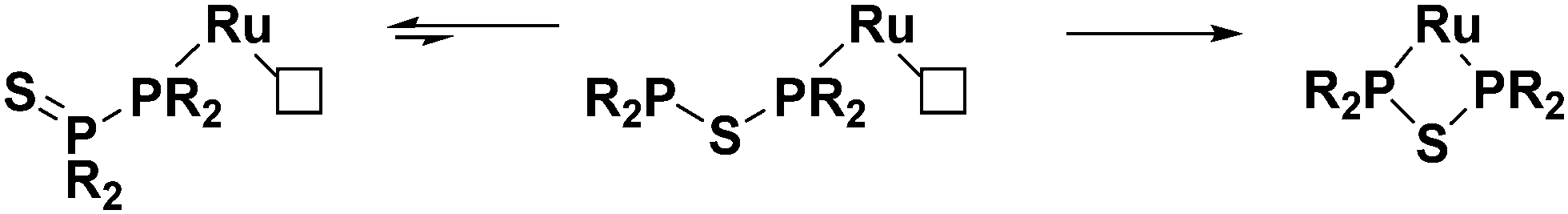 | ||
| Fig. 5 Proposed mechanism for the formation of 3 and 4. | ||
Our group has recently reported the synthesis of ruthenium phosphido complexes with bidentate phosphine donors and their reactions with molecular oxygen.49,50 In an effort to generate similar phosphido species with a dppte ligand, attempts were made to replace the chloride in 3 with a secondary phosphine using a synthetic procedure that has been successfully employed for the synthesis of other phosphido products from analogous ruthenium starting materials.49 The method utilized stoichiometric amounts of AgOTf as a halogen abstracting agent, but in the dppte case, an excess of AgOTf was necessary to ensure the removal of all of the chloride ligand. This in turn required an excess of diphenylphosphine as the silver cations in solution competed with the ruthenium centre for the monodentate phosphine (see Fig. 6A). Separation of the silver and ruthenium species was not trivial and required several recrystallization steps, which led to unacceptable yields of the target complex (less than 10%).
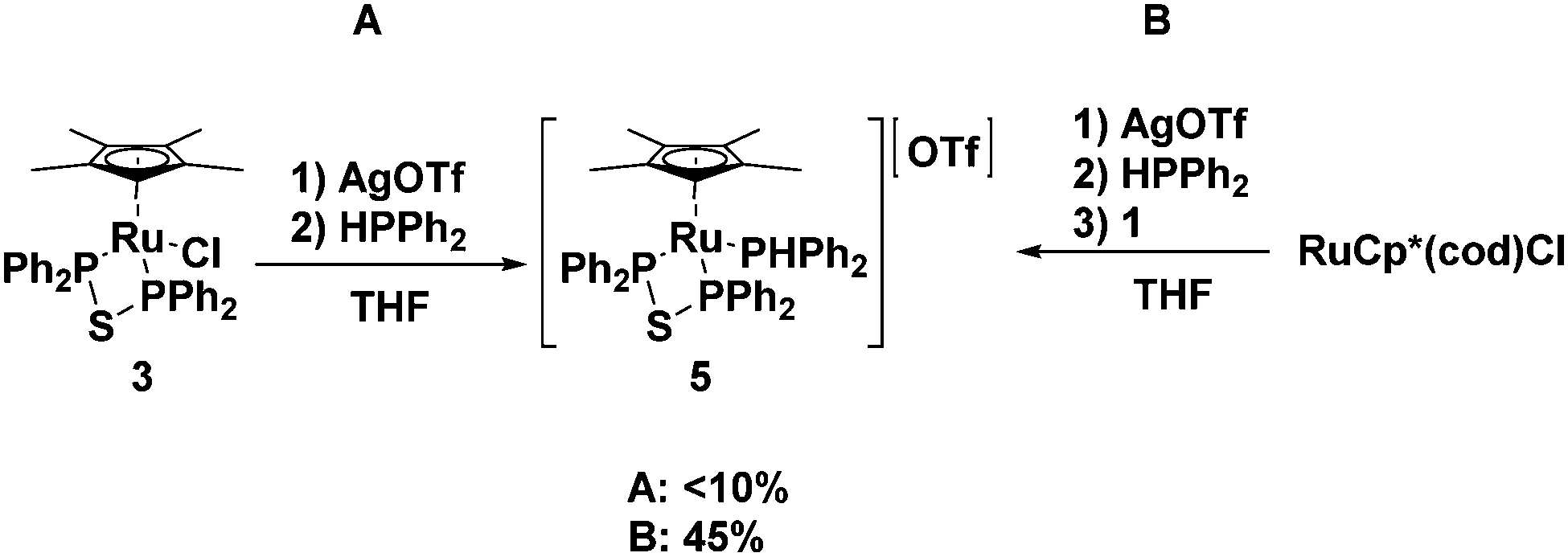 | ||
| Fig. 6 Two alternative routes to synthesizing complex 5, starting from 3 (left) or RuCp*(cod)Cl (right). | ||
In light of these poor results, an alternative synthetic scheme was developed exploiting RuCp*(cod)Cl as the starting material. In the first step, the chloride ligand was abstracted with AgOTf and diphenylphosphine was installed in its place (see Fig. 6B). A 31P NMR spectrum (decoupled) of the reaction mixture revealed two species in solution, at 31.6 and 30.5 ppm, both with a large P–H coupling, 374 and 347 Hz, respectively. Upon addition of 1, the signal at 31.6 ppm disappeared over time to give a new product, which showed a doublet, and a doublet of doublets at 32.4 and 29.4 ppm, respectively, while the other signal persisted in solution along with unreacted 1. The signal at 30.5 ppm has since been identified as [RuCp*(HPPh2)3][OTf], S1 (see ESI† for a crystal structure of S1 and a more detailed account of the synthesis of 5), which, based on our proposed mechanism for the formation of the bidentate ligand, explains why this species was inert to ligand substitution; the diphosphine monosulfide 1 was unable to displace a diphenylphosphine ligand. The doublet and doublet of doublets, on the other hand, were very diagnostic for the desired product: the doublet represented the equivalent phosphorus nuclei from the bidentate ligand, while the doublet of doublets corresponded to diphenylphosphine, which displayed a strong P–H coupling of 356 Hz (the P–H proton was also evident in the 1H NMR). Recrystallization of the reaction mixture allowed for separation of the desired product [RuCp*((PPh2)2S)(HPPh2)]OTf, 5, in poor yields (45%) as a yellow crystalline solid (see Fig. 6).
The XRD structure of 5 revealed a piano-stool structure with the bidentate and diphenylphosphine ligands cis to one another (see Fig. 7). The dppte ligand had a P(1)–Ru(1)–P(2) bite angle of 74.90(6)°, which is smaller than the bite angle seen in 3, but still larger than that of dppm.2 The P(1)–S(1)–P(2) bond angle, on the other hand, was found to be 82.46(9)° in 5, which is larger than that of 3, but still much smaller than the molybdenum carbonyl species and monometallic dppm complexes found in the literature (see Table S3 (ESI†) for other notable bond lengths and angles).2,41
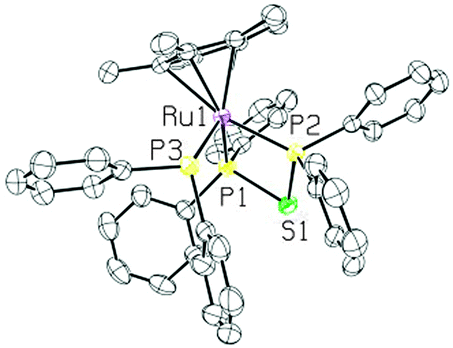 | ||
| Fig. 7 ORTEP3 representation and atom numbering for 5 (thermal ellipsoids at 50% probability; the solvent, counter ion and all the hydrogens are omitted for clarity). | ||
It should be noted that the 31P{1H} NMR spectrum of 5 varies significantly depending on the NMR solvent. Both the chemical shift and the JPP coupling constants change upon moving from deuterated THF to deuterated MeOH. Both sets of signals shift downfield to 36.3 and 34.4 ppm in the more polar solvent, for the doublet of doublets and the doublet, respectively. The Ph2PH phosphorus nucleus seems to be more affected by the different solvents, and the doublet of doublet shape becomes more apparent in MeOH-d4: JPP changes from 36 and 35 Hz to 37 and 33 Hz. The diphenylphosphine ligand produces a doublet of doublets rather than a triplet pattern because of residual coupling to the P–H proton due to the extremely high JPH (a decoupling problem).
With 5 in hand, deprotonation of the diphenylphosphine ligand was attempted in THF with an excess of KH. The reaction, however, did not proceed cleanly and did not yield the desired phosphido species. Deprotonation with KOtBu gave even poorer results and resulted in complete decomposition of the ruthenium dppte complex.
We have developed a simple and effective way of preparing alkyl- and aryl-substituted ligand precursors in the form of diphosphine monosulfides, and demonstrated that in the presence of a metal these compounds ‘open up’ to give the desired bidentate ligand. This valuable discovery makes a previously unattainable class of dppm-like ligands, with varied substituents on the phosphorus donors, readily available, even those thought to be inaccessible due to the instability of the free diphosphinothioether. In addition, we have characterized three metal complexes bearing these ligands and explored their stability with respect to different solvents and basic conditions. It was found that they tolerated a wide range of solvents, but were unstable in the presence of a strong base. More research is needed to explore the chemistry of this underutilized class of ligands.
This work was funded by NSERC Canada as Discovery and RTI grants to RHM.
Notes and references
- H. A. Mayer and W. C. Kaska, Chem. Rev., 1994, 94, 1239–1272 CrossRef CAS.
- R. J. Puddephatt, Chem. Soc. Rev., 1983, 12, 99–127 RSC.
- B. Chaudret, B. Delavaux and R. Poilblanc, Coord. Chem. Rev., 1988, 86, 191–243 CrossRef CAS.
- S. E. Kabir and G. Hogarth, Coord. Chem. Rev., 2009, 253, 1285–1315 CrossRef CAS.
- I. Ojima, C.-Y. Tsai, M. Tzamarioudaki and D. Bonafoux, Organic Reactions, John Wiley & Sons, Inc., 2004 Search PubMed.
- Z. Freixa and P. W. N. M. van Leeuwen, Dalton Trans., 2003, 1890–1901 RSC.
- P. W. N. M. van Leeuwen, P. C. J. Kamer and J. N. H. Reek, Pure Appl. Chem., 1999, 71, 1443–1452 CrossRef CAS.
- C. Krawiecki and J. Michalski, J. Chem. Soc., 1960, 881–885 RSC.
- J. B. Lambert, G. F. Jackson and D. C. Mueller, J. Am. Chem. Soc., 1970, 92, 3093–3097 CrossRef CAS.
- L. Maier, Helv. Chim. Acta, 1962, 45, 2381–2383 CrossRef CAS.
- L. Maier, J. Inorg. Nucl. Chem., 1962, 24, 275–283 CrossRef CAS.
- A. Marinetti and F. Mathey, Organometallics, 1982, 1, 1488–1492 CrossRef CAS.
- H. C. E. McFarlane, W. McFarlane and J. A. Nash, J. Chem. Soc., Dalton Trans., 1980, 240–244 RSC.
- E. N. Ofitserov, O. G. Sinyashin, E. S. Batyeva and A. N. Pudovik, J. Gen. Chem. USSR (Engl. Transl.), 1981, 51, 738–745 CAS.
- A. A. Pinkerton and R. G. Cavell, J. Am. Chem. Soc., 1972, 94, 1870–1874 CrossRef CAS.
- R. Schmutzler, O. Stelzer and N. Weferling, Chem. Ber., 1988, 121, 391–395 CrossRef CAS.
- M. Well and R. Schmutzler, Phosphorus, Sulfur Silicon Relat. Elem., 1992, 72, 171–187 CrossRef CAS.
- H. Westerman, M. Nieger and E. Niecke, Chem. Ber., 1991, 124, 13–16 CrossRef.
- M. Yoshifuji, K. Shibayama and N. Inamoto, Chem. Lett., 1984, 13, 115–118 CrossRef.
- E. W. Abel, D. A. Armitage and R. P. Bush, J. Chem. Soc., 1964, 5584–5587 RSC.
- T. Appleby and J. Derek Woollins, Coord. Chem. Rev., 2002, 235, 121–140 CrossRef CAS.
- D. E. J. Arnold, E. R. Cromie and D. W. H. Rankin, J. Chem. Soc., Dalton Trans., 1977, 1999–2004 RSC.
- D. J. Berg, R. A. Andersen and A. Zalkin, Organometallics, 1988, 7, 1858–1863 CrossRef CAS.
- P. Bhattacharyya, A. M. Z. Slawin, M. B. Smith, D. J. Williams and J. D. Woollins, J. Chem. Soc., Dalton Trans., 1996, 3647–3651 RSC.
- A. B. Burg and R. A. Sinclair, J. Am. Chem. Soc., 1966, 88, 5354–5355 CrossRef CAS.
- A. B. Burg and D.-K. Kang, J. Am. Chem. Soc., 1970, 92, 1901–1908 CrossRef CAS.
- A. B. Burg and K. Gosling, J. Am. Chem. Soc., 1965, 87, 2113–2116 CrossRef CAS.
- R. G. Cavell and H. J. Emeleus, J. Chem. Soc., 1964, 5825–5832 RSC.
- R. G. Cavell and A. A. Pinkerton, J. Am. Chem. Soc., 1971, 93, 2384–2390 CrossRef CAS.
- R. G. Cavell, R. D. Leary, A. R. Sanger and A. J. Tomlinson, Inorg. Chem., 1973, 12, 1374–1380 CrossRef CAS.
- R. C. Dobbie, L. F. Doty and R. G. Cavell, J. Am. Chem. Soc., 1968, 90, 2015–2020 CrossRef CAS.
- R. C. Dobbie and M. J. Hopkinson, J. Fluorine Chem., 1974, 3, 367–374 CrossRef.
- L. F. Doty and R. G. Cavell, Inorg. Chem., 1974, 13, 2722–2729 CrossRef CAS.
- J. A. S. Duncan, E. A. V. Ebsworth, R. O. Gould, C. L. Jones, D. W. H. Rankin and J. D. Whitelock, J. Chem. Soc., Dalton Trans., 1981, 1028–1033 RSC.
- M. A. El-Hinnawi, E. J. Wucherer, R. Lal De and H. Vahrenkamp, J. Organomet. Chem., 1987, 329, 381–390 CrossRef CAS.
- V. L. Foss, P. L. Kukhimisterov and I. F. Lutsenko, J. Gen. Chem. USSR (Engl. Transl.), 1982, 52, 1054–1062 CAS.
- K. Gosling and A. B. Burg, J. Am. Chem. Soc., 1968, 90, 2011–2015 CrossRef CAS.
- G. M. Gray and C. S. Kraihanzel, J. Organomet. Chem., 1982, 238, 209–222 CrossRef CAS.
- M. Gruber, P. G. Jones and R. Schmutzler, Chem. Ber., 1990, 123, 1313–1317 CrossRef CAS.
- M. Gruber and R. Schmutzler, Phosphorus, Sulfur Silicon Relat. Elem., 1993, 80, 181–194 CrossRef CAS.
- H. Einspahr and J. Donohue, Inorg. Chem., 1974, 13, 1839–1843 CrossRef CAS.
- M. Scheer, F. Uhlig, T. T. Nam, M. Dargatz, H. D. Schädler and E. Herrmann, Z. Anorg. Allg. Chem., 1990, 585, 177–188 CrossRef CAS.
- F. A. Cotton, L. R. Falvello, M. Tomas, G. M. Gray and C. S. Kraihanzel, Inorg. Chim. Acta, 1984, 82, 129–139 CrossRef CAS.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- P. W. N. M. van Leeuwen, P. C. J. Kamer, C. Claver, O. Pàmies and M. Diéguez, Chem. Rev., 2011, 111, 2077–2118 CrossRef CAS PubMed.
- B. R. Aluri, S. Peitz, A. Wöhl, N. Peulecke, B. H. Müller, A. Spannenberg and U. Rosenthal, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o404 CAS.
- P. G. Jones, A. K. Fischer, M. Farkens and R. Schmutzler, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, m478–m479 CAS.
- M. A. Pudovik, L. K. Kibardina, I. A. Aleksandrova, V. K. Khairullin and A. N. Pudovik, J. Gen. Chem. USSR, 1981, 51, 530–538 CAS.
- P. E. Sues, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2014 DOI:10.1021/ja5006724.
- P. E. Sues, M. W. Forbes, A. J. Lough and R. H. Morris, Dalton Trans., 2014, 43, 4137–4145 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section. Crystal structure and discussion of 1. Crystal structure and discussion of S1. Selected bond lengths and angles for 1, 2, 3, 4, and 5. Crystallographic data tables for compounds 1, 2, 3, 4, 5, and S1. CCDC 976592–976597. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc49456j |
| This journal is © The Royal Society of Chemistry 2014 |