 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Well-defined molecular uranium(III) chloride complexes†
Henry S.
La Pierre
,
Frank W.
Heinemann
and
Karsten
Meyer
*
Department of Chemistry and Pharmacy, Inorganic Chemistry, Friedrich-Alexander University Erlangen-Nürnberg (FAU), Egerlandstraße 1, 91058 Erlangen, Germany. E-mail: karsten.meyer@fau.de
First published on 21st February 2014
Abstract
The first anhydrous molecular complexes of uranium(III) chloride, soluble in polar aprotic solvents, are reported, including the structures of the dimeric [UCl3(py)4]2 and the trimetallic [UCl(py)4(μ-Cl)3U(py)2(μ-Cl)3UCl2(py)3].
The binary salt, UCl3, was first prepared by Péligot in 1842 en route to his landmark isolation of uranium metal.1 His synthesis required reducing UCl4 at 500 °C with dihydrogen. Despite the central importance of transition, main group, and f-block metal chlorides as starting materials in inorganic and organometallic synthesis, no simple procedure for the synthesis of a well-defined, anhydrous,2,3 and polar aprotic solvent soluble UCl3 starting material has yet been reported. While in situ methods for the reduction of UCl4 to UCl3 have been developed, the indeterminate nature of the U(III) species and the introduction of by-products that are potentially incompatible with subsequent reactions or efficient work-up have prevented their widespread adoption.4–6 In fact, the recent growth of trivalent uranium chemistry can be directly linked to the ground-breaking, robust methods for the synthesis of well-defined, soluble, molecular UBr3 and UI3 complexes, originally developed by Sattelberger and Clark.7–15
The development of protocols for the preparation of well-defined UCl3 precursors provides an alternative halide partner in salt metathesis reactions that produce more readily removed salt by-products, especially when the transmetallation reagents are limited to lithium salts. Additionally, it has been noted that the difficulty in removing alkali metal iodide salts can cause problems in subsequent synthetic procedures due to the propensity of I− to act as a reductant toward uranium(V) and (VI) species.16 Chloride is also a much more efficient bridging ligand due to its less nephelauxetic character than bromide or iodide. Therefore, UCl3 precursors may be efficient starting materials for building clusters via hydrolysis17–19 – necessary for modelling uranium waste stream products and biological waste remediation chemistry.20,21 Most importantly, from a synthetic perspective, uranium metal is not readily available outside the United States. The development of methods to access trivalent uranium from natural abundance or depleted UO3 (the immediate precursor to UCl4)10 allows for the ongoing research efforts of groups outside of the US. Herein, we report straightforward methods for the synthesis of [UCl3(py)4]2 and [UCl(py)4(μ-Cl)3U(py)2(μ-Cl)3UCl2(py)3].
The mild reducing conditions employed for the synthesis of [MoCl3(THF)3] from [MoCl4(THF)2], either with Zn or Sn metal at room temperature,22,23 suggested that a similar strategy may be applicable for the synthesis of a solvated UCl3 complex. Initial studies indicated that while Sn shot led to no reduction of [UCl4] in 1,4-dioxane at room temperature, Mg metal leads to the gradual (2 weeks) conversion of green [UCl4] to a blue suspension. For comparison, Kiplinger has recently reported the synthesis of the uranium(IV) dimer [UCl4(1,4-dioxane)]2 as a yellow/orange powder.24 Since [MgCl2(1,4-dioxane)2] is also insoluble in 1,4-dioxane, direct identification of the product is not possible, but elemental analysis suggests the formula [UCl3(1,4-dioxane)2]2·[MgCl2(1,4-dioxane)2]. Identification and purification of the solvated UCl3 were performed by dissolution in pyridine to give a deep purple solution, followed by fractional crystallization from Et2O/pyridine at −35 °C.
Optimization of this procedure was achieved by heating the 1,4-dioxane suspension of [UCl4] and Mg turnings to 100 °C for 3 days (Scheme 1). The blue suspension can be separated from the remaining Mg turnings by decantation onto a glass frit. Dissolution of the blue powder in pyridine (∼25 mL per 400 mg of UCl4), followed by filtration to remove a fine, pale green powder (may be the result of ether cleavage or incomplete reduction; however, the identity of this side-product is unknown), and precipitation with 1.4 equivalents of hexane (v/v of pyridine) lead to the isolation of [UCl3(py)4]2, 1, in 72% yield as small purple/black crystals (1 is vacuum sensitive and pyridine incorporation should be confirmed by elemental analysis, the box-scale procedure leads to the isolation of 1 in 41% yield and precisely four equivalents of py per U). The UV/vis/NIR spectrum (Fig. 1, black trace) of a 0.01 M solution of 1 clearly indicates trivalent uranium and the absence of tetravalent uranium. The spectrum of 1 is quite similar to that reported for the in situ prepared “UCl3(THF)x” in THF.41H NMR spectroscopy of 1 in pyridine-d5 with a hexamethylbenzene standard confirms the formula ratio of four pyridines per uranium. However, due to rapid solvent exchange, no peaks associated with bound pyridine can be observed. Most importantly, complex 1 can serve as a synthetic precursor. Hence, the reaction of 1 with 3.0 equivalents of NaN(SiMe3)2 in THF affords the previously reported [U(N(SiMe3)2)3(py)] in 78% yield.25
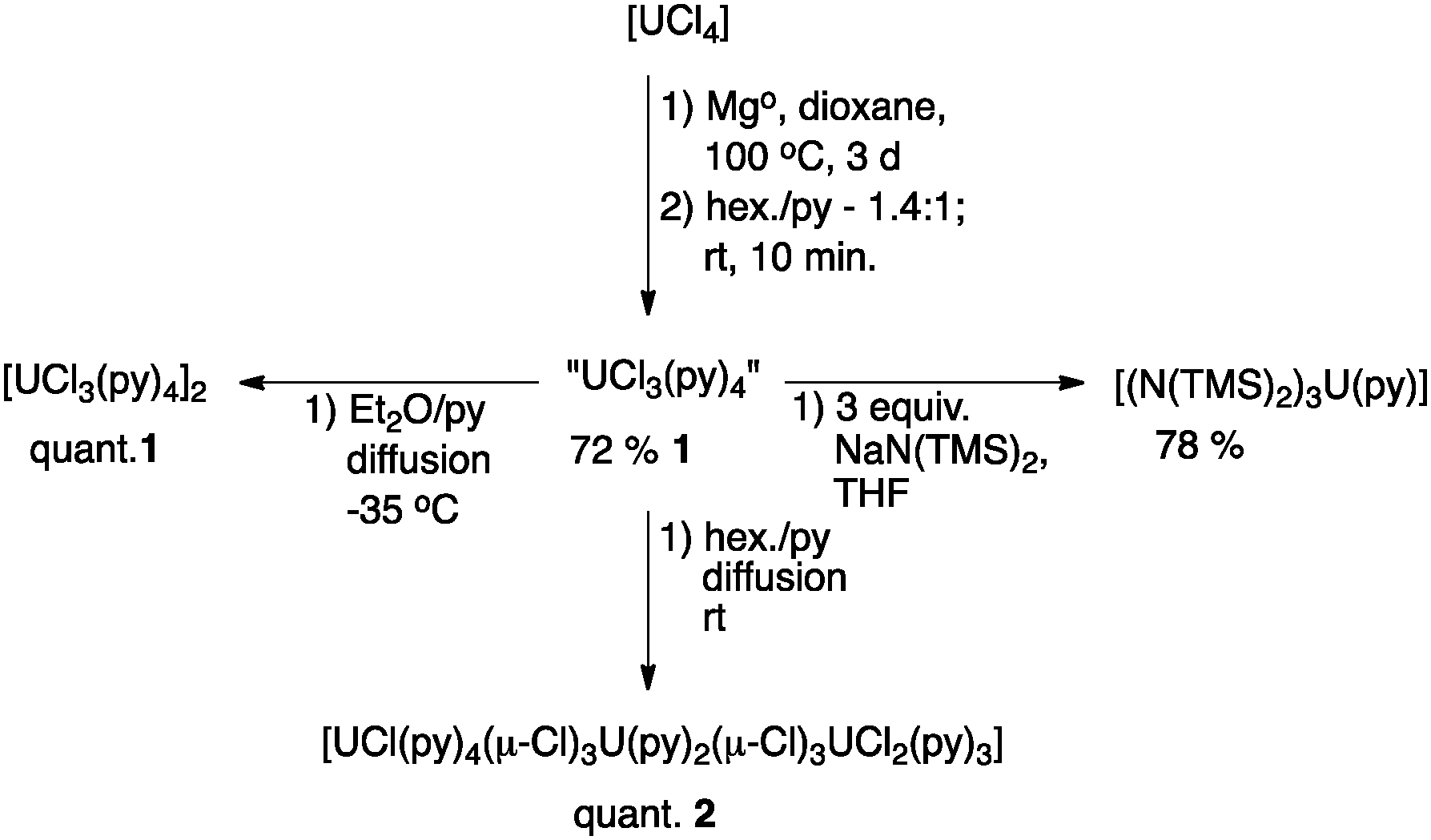 | ||
| Scheme 1 Synthetic procedures for [UCl3(py)4]2 and [UCl(py)4(μ-Cl)3U(py)2(μ-Cl)3UCl2(py)3]. | ||
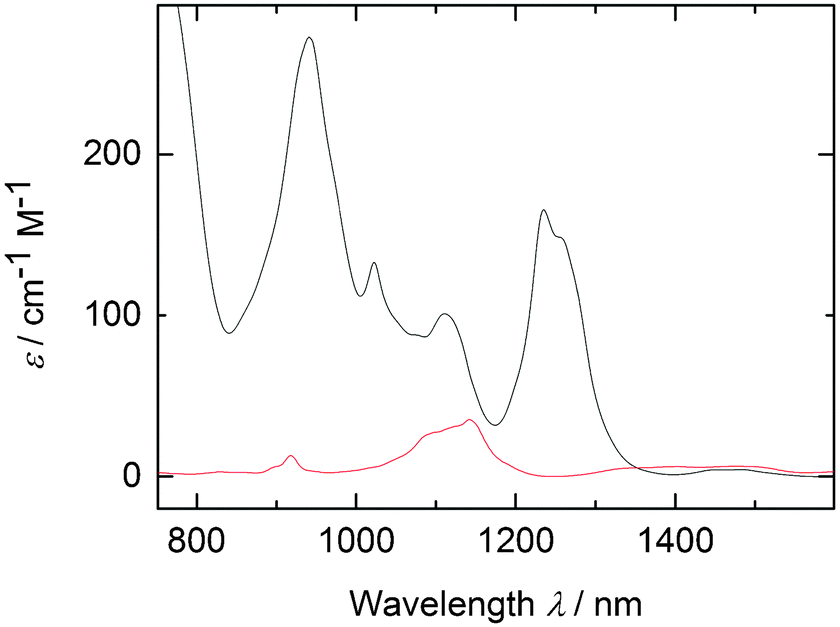 | ||
| Fig. 1 Absorption spectra of [UCl3(py)4], 0.01 M in py (black), and of [UCl4], 0.01 M in THF (red), [UCl4] is poorly soluble in py. | ||
 | ||
| Fig. 2 Molecular structures of 1 (in crystals of 1·4C5H5N) and of 2. Thermal ellipsoids set at 50% probability. Hydrogen atoms and co-crystallized solvent are removed for clarity; U: pink, Cl: green, N: blue, C: dark-grey. | ||
The solid-state molecular structure of 1 was determined by an X-ray diffraction study of crystals 1·4C5H5N grown by the diffusion of diethyl ether into a pyridine solution of 1 at −35 °C (Fig. 2). Crystals prepared in this manner grow as thin layered plates necessitating the acquisition of data on two layers and the solution of the structure as a non-merohedral twin. This solution reveals 1 to be a chloride-bridged dimer in the solid-state with a U2Cl2 diamond core structural motif. The U⋯U distance is 4.71 Å and the U–Cl–U angle is 108.9°. The bridging U–Cl distances are 2.935(2) and 2.853(2) Å (2.936(2) and 2.862(2) Å) (two independently refined UCl3(py)4 fragments per asymmetric unit), whereas the terminal U–Cl bond lengths are 2.734(2), 2.759(2), 2.740(2), and 2.768(2) Å. The U–N distances span the range from 2.690(8) to 2.772(7) Å.
In an attempt to grow crystals of 1 in another lattice setting, another product, namely [UCl(py)4(μ-Cl)3U(py)2(μ-Cl)3UCl2(py)3], 2, was observed in quantitative yield when crystals were prepared by the diffusion of hexane into a dilute solution of 1 in pyridine at room temperature. Notably, to the best of our knowledge, there is no precedent for a trimetallic complex of uranium(III) bridged by a (μ-Cl)3 unit. In complex 2, the U⋯U distances are inequivalent at 4.061 Å (U1⋯U2) and 4.176 Å (U2⋯U3), respectively. The bridging U–Cl bond distances for U1⋯U2 are shorter and U1–Cl–U2 angles are contracted in comparison to those for U2⋯U3. Specifically, the bridging U–Cl bond distance for U1⋯U2 averages to 2.88 Å and 2.89 Å for U2⋯U3. Similarly, the U1–Cl–U2 angles span 88.68(2) to 90.57(2)°, while the U2–Cl–U3 angles span from 92.19(3) to 92.71(3)°. A similar trimetallic complex of uranium(IV) was presented by Cotton in 1986,2,26 namely [(η6-C6Me6)UCl2(μ-Cl)3UCl2(μ-Cl)3UCl2(η6-C6Me6)]. The U–Cl bond distances are considerably shorter (2.666(7) to 2.762(6) Å) and the U–Cl–U angles range from 92.4(2) to 93.6(2)° to yield similar U⋯U distances to those found in 2 (4.035(1) and 4.031(1) Å). However, in comparison to 2, the central uranium ion is equidistant from the other two uranium ions. The asymmetric bridging (μ-Cl)3 moiety in 2 may be due to the different pyridine binding in U3 and U1 and crystal packing forces. However, the potential for antiferromagnetic coupling to perturb the ground state geometry cannot be discounted.
In conclusion, the first well-defined molecular complexes of uranium trichloride with practical solubility in common laboratory solvents were prepared and characterized. Reduction of [UCl4] with magnesium turnings affords [UCl3(py)4] readily. Recrystallization from Et2O/pyridine affords the dimeric [UCl3(py)4]2 and recrystallization from hexane/pyridine yields the trimetallic complex, [UCl(py)4(μ-Cl)3U(py)2(μ-Cl)3UCl2(py)3]. These compounds should be widely employable reagents for further developments in uranium(III) inorganic coordination and organometallic chemistry. Magnetism studies on the unique trimetallic uranium(III) complex, [UCl(py)4(μ-Cl)3U(py)2(μ-Cl)3UCl2(py)3], 2, are on-going.
Financial support was provided the Bundesministerium für Bildung und Forschung (BMBF 2020+, 02NUK012B), the FAU Erlangen-Nuremberg, and COST Action CM1006. The authors thank Dr. Regine Herbst-Irmer (Georg-August-Universität Göttingen) for a helpful discussion of the twinning issue.
Notes and references
- E.-M. Péligot, Ann. Chim. Phys., 1842, 5, 5 Search PubMed.
- J. Drożdżyński, Inorg. Chim. Acta, 1985, 109, 79 CrossRef.
- A. Mech, M. Karbowiak, T. Lis and J. Drozdzynski, Polyhedron, 2006, 25, 2083 CrossRef CAS PubMed.
- D. C. Moody and J. D. Odom, J. Inorg. Nucl. Chem., 1979, 41, 533 CrossRef CAS.
- R. A. Andersen, Inorg. Chem., 1979, 18, 1507 CrossRef CAS.
- J. MacCordick and C. Brun, C. R. Chim., 1970, 270, 620 CAS.
- D. L. Clark, A. P. Sattelberger, S. G. Bott and R. N. Vrtis, Inorg. Chem., 1989, 28, 1771 CrossRef CAS.
- L. R. Avens, S. G. Bott, D. L. Clark, A. P. Sattelberger, J. G. Watkin and B. D. Zwick, Inorg. Chem., 1994, 33, 2248 CrossRef CAS.
- I. A. Khan and H. S. Ahuja, Inorg. Synth., 1982, 21, 187 CrossRef CAS.
- J. L. Kiplinger, D. E. Morris, B. L. Scott and C. J. Burns, Organometallics, 2002, 21, 5978 CrossRef CAS.
- F. G. N. Cloke and P. B. Hitchcock, J. Am. Chem. Soc., 2002, 124, 9352 CrossRef CAS PubMed.
- W. J. Evans, S. A. Kozimor, J. W. Ziller, A. A. Fagin and M. N. Bochkarev, Inorg. Chem., 2005, 44, 3993 CrossRef CAS PubMed.
- C. D. Carmichael, N. A. Jones and P. L. Arnold, Inorg. Chem., 2008, 47, 8577 CrossRef CAS PubMed.
- D. D. Schnaars, G. Wu and T. W. Hayton, Dalton Trans., 2008, 6121 RSC.
- L. Natrajan, M. Mazzanti, J. P. Bezombes and J. Pecaut, Inorg. Chem., 2005, 44, 6115 CrossRef CAS PubMed.
- S. Fortier, J. L. Brown, N. Kaltsoyannis, G. Wu and T. W. Hayton, Inorg. Chem., 2012, 51, 1625 CrossRef CAS PubMed.
- L. Natrajan, F. Burdet, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2006, 128, 7152 CrossRef CAS PubMed.
- G. Nocton, F. Burdet, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed., 2007, 46, 7574 CrossRef CAS PubMed.
- B. Biswas, V. Mougel, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed., 2011, 50, 5744 Search PubMed.
- Y. Suzuki, S. D. Kelly, K. M. Kemner and J. F. Banfield, Nature, 2002, 419, 134 CrossRef CAS PubMed.
- B. H. Gu, H. Yan, P. Zhou, D. B. Watson, M. Park and J. Istok, Environ. Sci. Technol., 2005, 39, 5268 CrossRef CAS.
- J. R. Dilworth and J. Zubieta, Inorg. Synth., 1986, 24, 193 CrossRef CAS.
- J. R. Dilworth and R. L. Richards, Inorg. Synth., 1980, 20, 121 CrossRef.
- M. J. Monreal, R. K. Thomson, T. Cantat, N. E. Travia, B. L. Scott and J. L. Kiplinger, Organometallics, 2011, 30, 2031 CrossRef CAS.
- J. L. Stewart, PhD thesis, University of California, Berkeley, CA, 1988.
- G. C. Campbell, F. A. Cotton, J. F. Haw and W. Schwotzer, Organometallics, 1986, 5, 274 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and crystallographic data (PDF and CIF). CCDC 976568 (1) and 976569 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc49452g |
| This journal is © The Royal Society of Chemistry 2014 |