 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly regioselective and chemoselective titanocene mediated Barbier-type allylation reactions†
Sara P.
Morcillo
a,
Ángela
Martínez-Peragón
a,
Verena
Jakoby
b,
Antonio J.
Mota
c,
Christian
Kube
b,
José
Justicia
a,
Juan M.
Cuerva
*a and
Andreas
Gansäuer
*b
aDepartment of Organic Chemistry, University of Granada, Fuentenueva Campus, Granada 18071, Spain. E-mail: jmcuerva@ugr.es
bKekulé-Institut für Organische Chemie und Biochemie, Universität Bonn, Gerhard-Domagk-Strasse 1, 53121 Bonn, Germany. E-mail: andreas.gansaeuer@uni-bonn.de
cDepartment of Inorganic Chemistry, University of Granada, Fuentenueva Campus, Granada 18071, Spain
First published on 6th January 2014
Abstract
Titanocene carboxylate 1 is an excellent chemoselective reagent for unprecedented α-regioselective Barbier-type reactions. It constitutes the first titanocene(III) able to tolerate epoxides and readily reduced carbonyl compounds, such as aromatic and α,β-unsaturated aldehydes.
Titanocene(III) complexes are useful tools for mediating and catalyzing a number of useful transformations,1 such as homolytic epoxide opening,2 pinacol coupling reactions,3 and Barbier-type additions of allylic and propargylic halides or carbonates to carbonyl compounds.4 A drawback of these procedures is the lack of chemoselectivity in the electron transfer step. For this reason, aromatic and α,β-unsaturated carbonyl compounds are usually unsuitable substrates in Barbier-type reactions.4 Here, we demonstrate that this shortcoming can be resolved in the case of complex 1 (Scheme 1) in the presence of Mn dust via complex 2a (Fig. 1).5,6
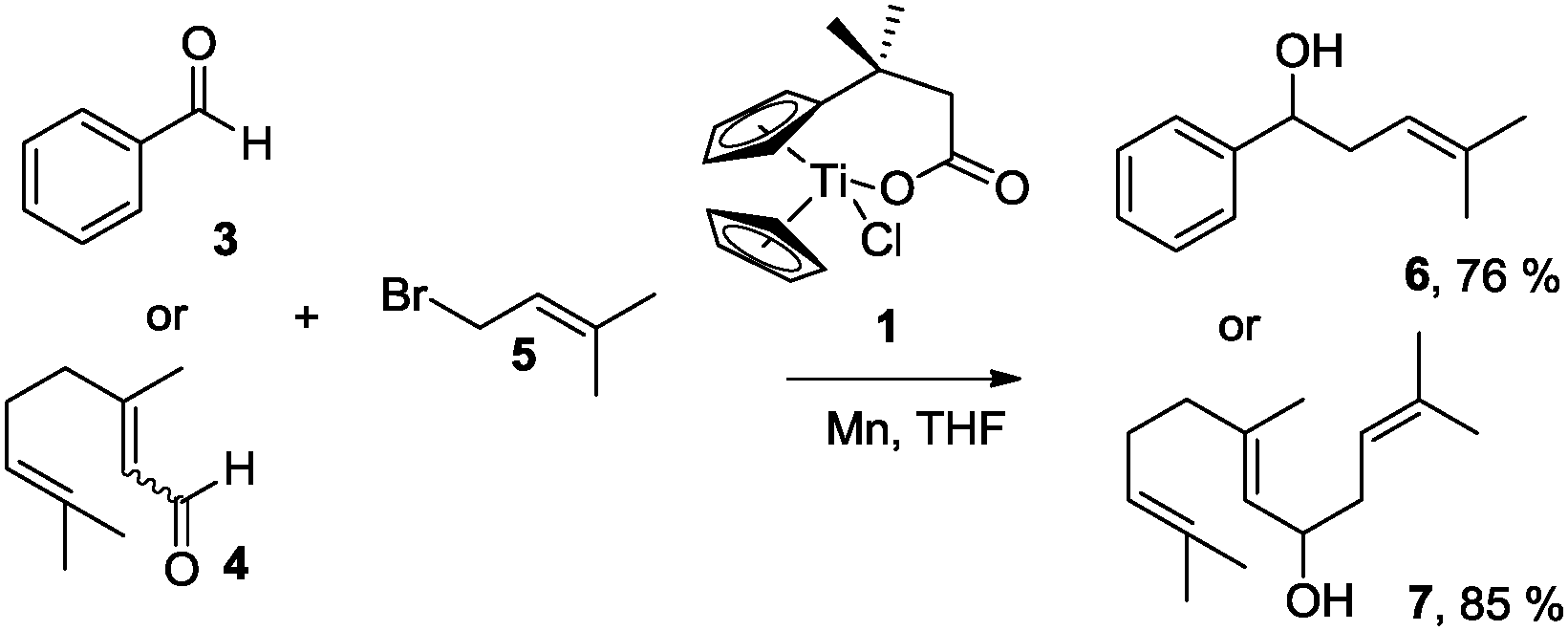 | ||
| Scheme 1 Ti-mediated regioselective α-prenylation of benzaldehyde (3) and citral (4). | ||
 | ||
| Fig. 1 CV of 2 mM solution of 1 (red) and Mn–1 (black) recorded at ν = 100 mV s−1 in 0.2 M TBAPF6–THF. | ||
Gratifyingly, 3 and 4, model compounds for aromatic and α,β-unsaturated aldehydes, reacted with 5 in the presence of complex 1 and Mn dust to exclusively yield α-prenylated compounds 6 and 7. Thus, the highly useful α-regioselectivity of prenylation noted earlier could be maintained.4a,6 Remarkably, in the absence of 5, no pinacol products are formed.7 Further exploring this new reactivity concluded that epoxides were also tolerated by this reagent. This suggests that the aldehydes or epoxides cannot bind to Mn–1.
To rationalize these findings, cyclic voltammograms (CVs) of 1 and Mn–1 in THF were recorded (Fig. 1). The oxidation of electrochemically reduced 1 reveals the presence of two species. 1− (Epc = −1.38 V vs. Fc+/Fc) is formed via electron transfer at the electrode. The process is irreversible due to the formation of 2a (Epa1 = −1.00 V) through loss of chloride. For Mn–1, 2a (Epa1 = −0.95 V) is formed as expected, due to the more efficient abstraction of the chloride and formation of MnCl2. The more negative value of Epa1 compared to Cp2TiCl (Epa1 = −0.83 V) is in line with the carboxylate that is a better donor ligand than chloride. The second peak (Epa2 = −0.65 V) is also observed at higher sweep rates (ν = 1–50 V s−1). Thus, the species being oxidized is an initial component8 of Mn–1 in THF and is not formed during the sweep. We suggest that 2b and not cationic 2c is the second component of Mn–1 for a number of reasons. 2c should have a potential similar to that of [Cp(C5H4tBu)Ti]+ (Epa = −0.47 V). It has been recently shown that cationic titanocene(III) complexes open epoxides.9 Both findings are in contradiction to the behavior of Mn–1. In agreement with the observations, 2b is more difficult to oxidize than 2a because the electron density is withdrawn from Ti through carboxylate coordination. The steric shielding of Ti in 2b will be similar to that in 2a and thus epoxide and aldehyde binding is as difficult for 2b as for 2a.

To understand the unprecedented chemoselectivity, we carried out the model reactions with Mn-reduced Cp2TiCl(OMe),10 Cp2TiCl(OAc),11 and Cp2TiCl(OBz).12 Unselective electron transfer reactions with Cp2TiCl2 were observed. This suggests that the tethering of the carboxylate to the ligand is essential.
To highlight this point, the coordination of 2a to THF, benzaldehyde, and trimethyloxirane was studied by DFT calculations (Table 1).7
|
|
|||
|---|---|---|---|
| Complex | ΔH | −TΔS | ΔG |
| 2a | 0.0 | 0.0 | 0.0 |
| 2a-THF | −2.8 | +13.7 | +10.9 |
| 2a-B | −4.2 | +12.5 | +8.3 |
| 2a-Ep | −0.5 | +12.5 | +12.0 |
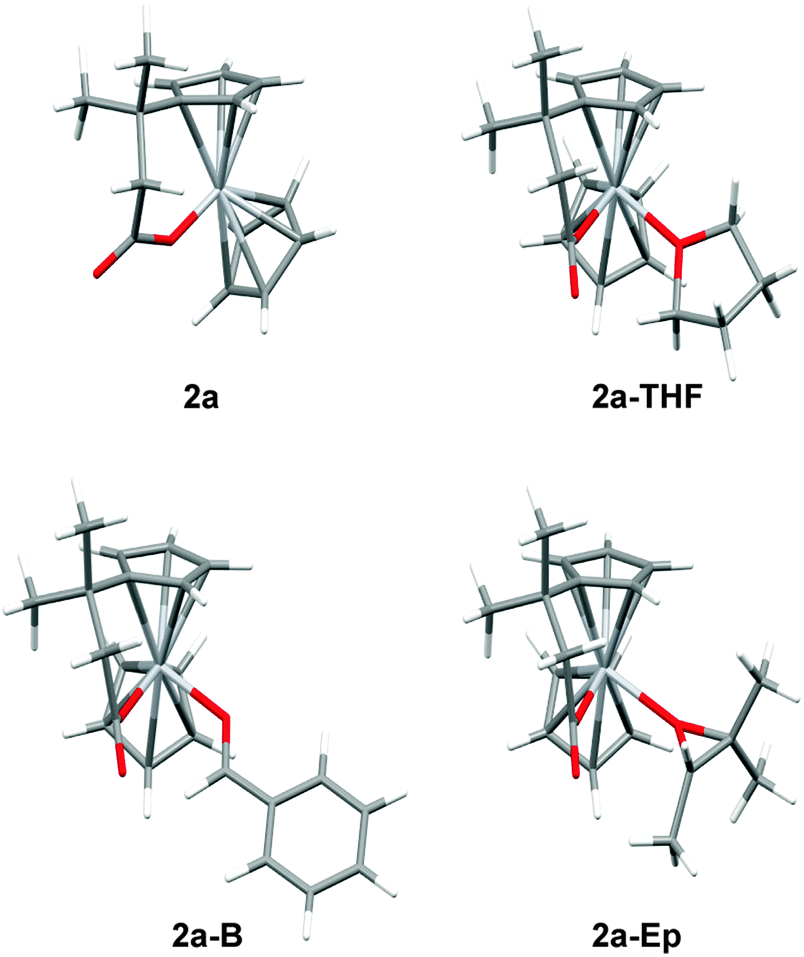
The generation of these coordination complexes is characterized by: (a) a only slightly negative enthaply of formation and (b) a highly unfavorable entropy of formation. This results in ΔG values of about +8 to +12 kcal mol−1.13 Therefore, aldehyde and epoxide complexation seems to be precluded as observed experimentally.
Next, we explored the chemoselectivity of electron transfer further (Scheme 2 and Table 2). Gratifyingly, our Barbier-type reactions using activated halides as pronucleophiles are general and take place with moderate to excellent yields.7 A variety of aromatic and α,β-unsaturated aldehydes are suitable. Even acetophenone, as an example of aromatic ketones, is an excellent substrate (Scheme 2). All products are valuable building blocks for terpene synthesis.1d Even more interesting and demanding are Barbier-type reactions with electrophiles (19–20) or nucleophiles (23), incorporating epoxides (Table 2).
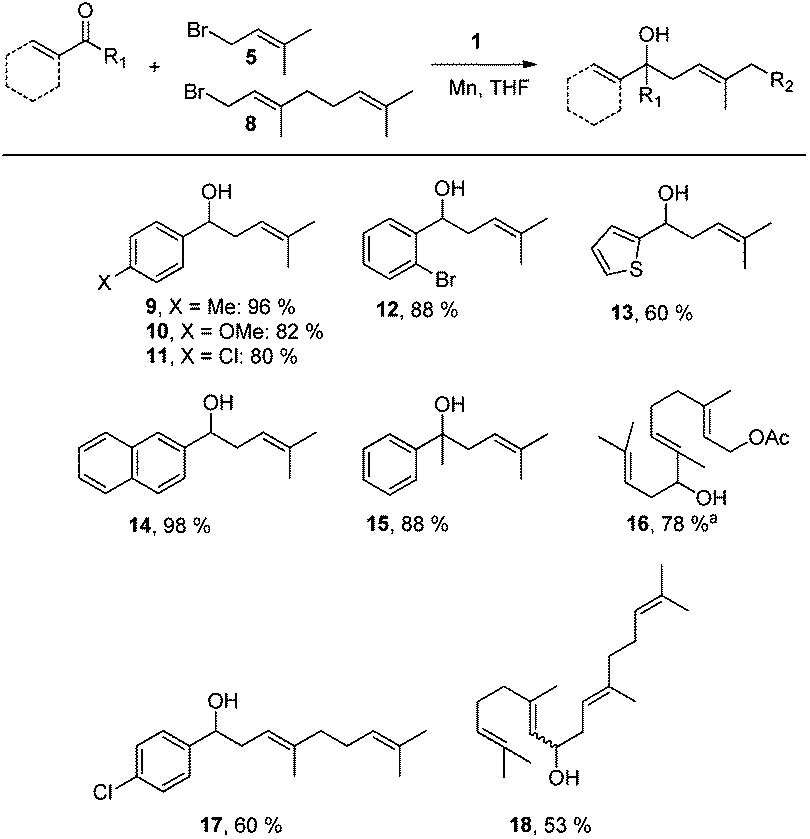 | ||
| Scheme 2 Ti-mediated regioselective α-prenylation of aromatic and α,β-unsaturated carbonyl compounds. Conditions: for aromatic aldehydes: 1 (1 mmol), Mn dust (8 mmol), aldehyde (1 mmol), and activated halide (2 mmol) in THF. Otherwise: 1 (1.5 mmol), Mn dust (8 mmol), carbonyl compound (1 mmol), and activated halide (3 mmol) in THF. aPrenyl chloride can be used with similar yield (77%). | ||
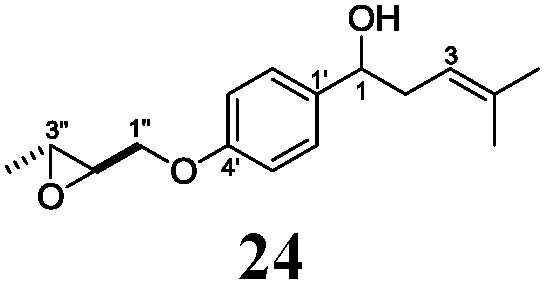
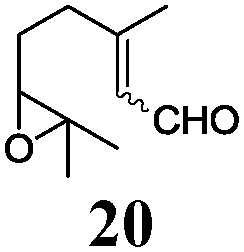
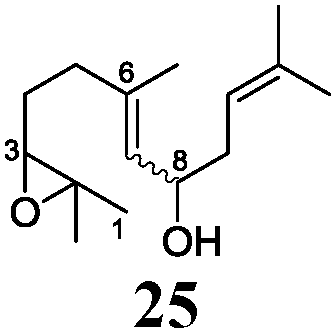
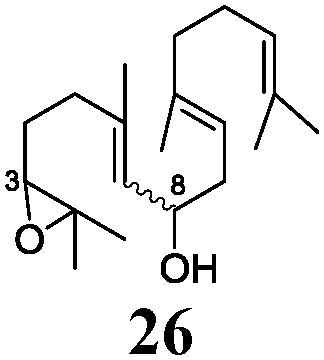

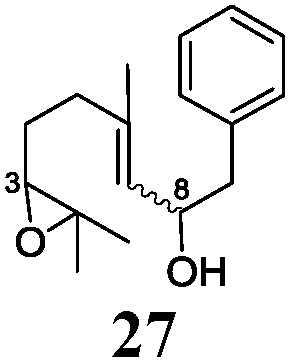

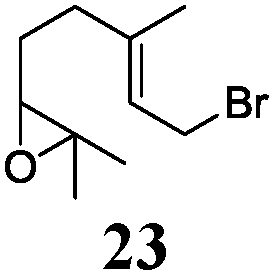
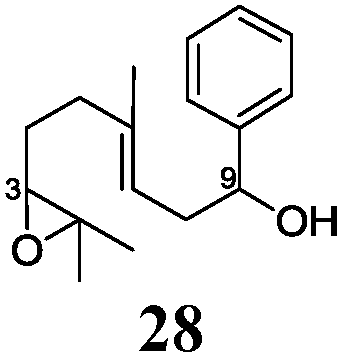
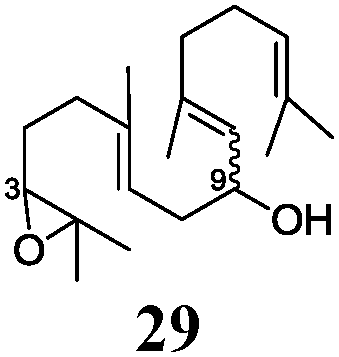
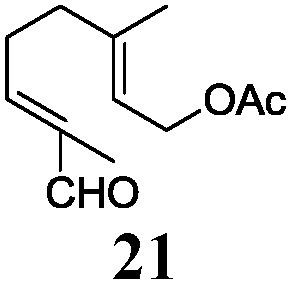
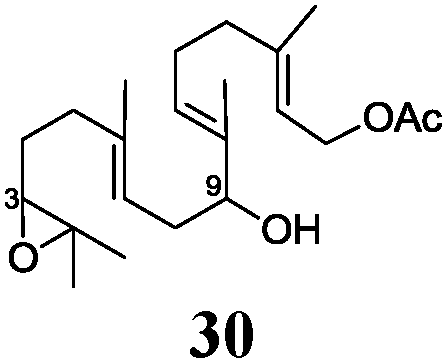
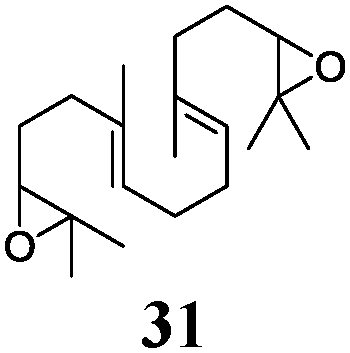
In agreement with our DFT calculations, epoxides are readily tolerated by Mn–1.7 Thus, functionalized epoxypolyprenes that are highly attractive intermediates for synthesis of natural products1d can be prepared from either epoxide containing carbonyl compounds or activated halides. As an attractive additional feature, our novel method allows the elimination of the often difficult regioselective epoxidation step of the polyprenic starting materials.
An important aspect of this study is the regioselectivity of the addition of the allylic nucleophile. In contrast to the numerous γ-selective additions, α-regioselective additions are considerably less common.14 Here, we obtained the α-regioisomers in very high selectivity (>92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :<8) when prenyl derivatives 5, 8 and 23 were used as pronucleophiles. Moreover, commercially available Mn dust could be used at room temperature.
:<8) when prenyl derivatives 5, 8 and 23 were used as pronucleophiles. Moreover, commercially available Mn dust could be used at room temperature.
Finally, we also investigated Wurtz-type coupling reactions mediated by Mn–1. Such reactions can be carried out in the presence of Cp2TiCl.15 However, the chemoselectivity of these reactions is low. This is not the case in our system and, hence, epoxide containing substrates can be readily coupled (Table 2, entry 8). The reaction took place with excellent yield and also with a high α,α-regioselectivity (86:14).
In summary, we have demonstrated that Mn–1 is a reagent for Barbier-type allylation reactions delivering the α-addition products with almost complete regioselectivity. The reactions proceed with an unprecedented chemoselectivity because epoxides and readily reduced carbonyl compounds, such as aromatic and α,β-unsaturated aldehydes, are tolerated. The compounds obtained are difficult to prepare using other methodologies and are valuable substrates in terpene synthesis. Moreover, the reactions can be performed at room temperature and no preformed organometallic reagents (B or Li) or especially activated metals (Ba) have to be employed.
We thank the Spanish MICINN (Grants CTQ-2011.22455 and PRI-AIBDE-2011-1122) and SFB 813 ‘Chemistry at Spin Centers’ for financial support. SPM thanks Regional Government of Andalucía for her contract. We also thank the Centro de Supercomputación de la Universidad de Granada (UGRGRID) for computational time.
Notes and references
- (a) A. Gansäuer, T. Lauterbach and S. Narayan, Angew. Chem., Int. Ed., 2003, 42, 5556–5573 CrossRef PubMed; (b) J. M. Cuerva, J. Justicia, J. L. Oller-López and J. E. Oltra, Top. Curr. Chem., 2006, 264, 63–91 CrossRef CAS; (c) A. Gansäuer, J. Justicia, C.-A. Fan, D. Worgull and F. Piestert, Top. Curr. Chem., 2007, 279, 25–52 CrossRef; (d) J. Justicia, L. Álvarez de Cienfuegos, A. G. Campaña, D. Miguel, V. Jakoby, A. Gansäuer and J. M. Cuerva, Chem. Soc. Rev., 2011, 40, 3525–3537 RSC.
- For seminal work, see: (a) T. V. RajanBabu and W. A. Nugent, J. Am. Chem. Soc., 1994, 116, 986–997 CrossRef CAS; (b) A. Gansäuer, H. Bluhm and M. Pierobon, J. Am. Chem. Soc., 1998, 120, 12849–12859 CrossRef.
- (a) Y. Handa and J. Inanaga, Tetrahedron Lett., 1987, 28, 5717–5718 CrossRef CAS; (b) A. Gansäuer, Chem. Commun., 1997, 457–458 RSC; (c) A. Gansäuer and D. Bauer, J. Org. Chem., 1998, 63, 2070–2071 CrossRef; (d) A. Gansäuer and D. Bauer, Eur. J. Org. Chem., 1998, 2673–2676 CrossRef; (e) T. Hirao, B. Hatano, M. Asahara, Y. Muguruma and A. Ogawa, Tetrahedron Lett., 1998, 39, 5247–5248 CrossRef CAS; (f) M. C. Barden and J. Schwartz, J. Am. Chem. Soc., 1996, 118, 5484–5485 CrossRef CAS; (g) M. Paradas, A. G. Campaña, R. E. Estévez, L. Álvarez de Cienfuegos, T. Jiménez, R. Robles, J. M. Cuerva and J. E. Oltra, J. Org. Chem., 2009, 74, 3616–3619 CrossRef CAS PubMed.
- (a) R. E. Estévez, J. Justicia, B. Bazdi, N. Fuentes, M. Paradas, D. Choquesillo-Lazarte, J. M. García-Ruiz, R. Robles, A. Gansäuer, J. M. Cuerva and J. E. Oltra, Chem.–Eur. J., 2009, 15, 2774–2791 CrossRef PubMed; (b) A. G. Campaña, B. Bazdi, N. Fuentes, R. Robles, J. M. Cuerva, J. E. Oltra, S. Porcel and A. M. Echavarren, Angew. Chem., Int. Ed., 2008, 47, 7515–7519 CrossRef PubMed; (c) A. Millán, L. Álvarez de Cienfuegos, A. Martín-Lasanta, A. G. Campaña and J. M. Cuerva, Adv. Synth. Catal., 2011, 353, 73–78 CrossRef; (d) A. Martínez-Peragón, A. Millán, A. G. Campaña, I. Rodríguez-Márquez, S. Resa, D. Miguel, L. Álvarez de Cienfuegos and J. M. Cuerva, Eur. J. Org. Chem., 2012, 1499–1503 CrossRef; (e) J. Muñoz-Bascón, I. Sancho-Sanz, E. Álvarez-Manzaneda, A. Rosales and J. E. Oltra, Chem.–Eur. J., 2012, 18, 14479–14486 CrossRef PubMed.
- (a) A. Gansäuer, D. Franke, T. Lauterbach and M. Nieger, J. Am. Chem. Soc., 2005, 127, 11622–11623 CrossRef PubMed; (b) A. Gansäuer, I. Winkler, D. Worgull, D. Franke, T. Lauterbach, A. Okkel and M. Nieger, Organometallics, 2008, 27, 5699–5707 CrossRef.
- T. Jiménez, S. P. Morcillo, A. Martín-Lasanta, D. Collado-Sanz, D. J. Cárdenas, A. Gansäuer, J. Justicia and J. M. Cuerva, Chem.–Eur. J., 2012, 18, 12825–12833 CrossRef PubMed.
- See ESI† for details.
- (a) R. J. Enemærke, G. H. Hjøllund, K. Daasbjerg and T. Skrydstrup, C. R. Acad. Sci., 2001, 4, 435–438 Search PubMed; (b) R. J. Enemærke, J. Larsen, T. Skrydstrup and K. Daasbjerg, Organometallics, 2004, 23, 1866–1874 CrossRef; (c) R. J. Enemærke, J. Larsen, T. Skrydstrup and K. Daasbjerg, J. Am. Chem. Soc., 2004, 126, 7853–7864 CrossRef PubMed; (d) R. J. Enemærke, J. Larsen, G. H. Hjøllund, T. Skrydstrup and K. Daasbjerg, Organometallics, 2005, 24, 1252–1262 CrossRef; (e) J. Larsen, R. J. Enemærke, T. Skrydstrup and K. Daasbjerg, Organometallics, 2006, 25, 2031–2036 CrossRef CAS; (f) A. Gansäuer, A. Barchuk, F. Keller, M. Schmitt, S. Grimme, M. Gerenkamp, C. Mück-Lichtenfeld, K. Daasbjerg and H. Svith, J. Am. Chem. Soc., 2007, 129, 1359–1371 CrossRef PubMed; (g) A. Gansäuer, K. Knebel, C. Kube, M. van Gastel, A. Cangönül, K. Daasbjerg, T. Hangele, M. Hülsen, M. Dolg and J. Friedrich, Chem.–Eur. J., 2012, 18, 2591–2599 CrossRef PubMed.
- A. Cangönül, M. Behlendorf, A. Gansäuer and M. van Gastel, Inorg. Chem., 2013, 52, 11859–11866 CrossRef PubMed.
- D. H. Gibson, Y. Ding, M. S. Mashuta and J. F. Richardson, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 559–560 CrossRef CAS.
- V. A. Knizhnikov, V. L. Shirokii and Y. A. Oldekop, Vestsi Akad. Navuk BSSR, Ser. Khim. Navuk, 1983, 3, 102–104 Search PubMed.
- B. S. Kaliral, J.-D. Foulon, T. A. Hamor, C. J. Jones, P. D. Beer and S. P. Fricker, Polyhedron, 1991, 10, 1847–1856 CrossRef.
- A direct comparison of the binding capabilities of complex 2a with parent Cp2TiCl is not simple owing to that it is known that the chloride anion dissociates in THF solution generating cationic species (ref. 9). Such an effect cannot take place with a tethering ligand in complex 2. Related zwitterionic species 2c was obtained and the results showed that in the absence of a ligand (L) the zwitterion is too high in energy to be a real intermediate, as CV experiments suggest. In the presence of a ligand, cationic complex 2c-L was found to be thermodynamically highly disfavoured species.
- (a) P. Girard, J. L. Namy and H. B. Kagan, J. Am. Chem. Soc., 1980, 102, 2693–2698 CrossRef CAS; (b) B.-S. Guo, W. Doubleday and T. Cohen, J. Am. Chem. Soc., 1987, 109, 4710–4711 CrossRef CAS; (c) A. Yanagisawa, S. Habaue, K. Yasue and H. Yamamoto, J. Am. Chem. Soc., 1994, 116, 6130–6141 CrossRef CAS; (d) S. Matsukawa, Y. Funabashi and T. Imamoto, Tetrahedron Lett., 2003, 44, 1007–1010 CrossRef CAS; (e) K. M. Depew, S. J. Danishefsky, N. Rosen and L. Sepp-Lorenzino, J. Am. Chem. Soc., 1996, 118, 12463–12464 CrossRef CAS; (f) B. Y. Park, T. P. Montgomery, V. J. Garza and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 16320–16323 CrossRef CAS PubMed.
- (a) A. F. Barrero, M. M. Herrador, J. F. Quilez del Moral, P. Arteaga, J. F. Arteaga, M. Piedra and E. M. Sanchez, Org. Lett., 2005, 7, 2301–2304 CrossRef CAS PubMed; (b) A. Millán, A. G. Campaña, B. Bazdi, D. Miguel, L. Álvarez de Cienfuegos, A. M. Echavarren and J. M. Cuerva, Chem.–Eur. J., 2011, 17, 3985–3994 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and characterisation of new compounds. See DOI: 10.1039/c3cc49230c |
| This journal is © The Royal Society of Chemistry 2014 |