 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning porosity and activity of microporous polymer network organocatalysts by co-polymerisation†
Johannes
Schmidt
*a,
Dipti Sankar
Kundu
b,
Siegfried
Blechert
*b and
Arne
Thomas
a
aTechnische Universität Berlin, Hardenbergstraße 40, 10623 Berlin, Germany. E-mail: johannes.schmidt@tu-berlin.de
bTechnische Universität Berlin, Straße des 17. Juni 135, 10623 Berlin, Germany. E-mail: blechert@chem.tu-berlin.de
First published on 10th February 2014
Abstract
Microporous polymer networks based on binaphthyl phosphoric acids are suitable heterogeneous asymmetric organocatalysts. Herein we show that the porous characteristics of such networks can be fine-tuned by co-polymerisation. This enables us to investigate the influence of the surface area and porosity in microporous networks on their catalytic performance. In this case, the activity of the polymers in an asymmetric hydrogenation reaction is increased by the use of polymers with higher surface areas.
Catalysis, one of the main fields of application of porous materials, has moved into the focus of microporous polymer research.2–6 In recent years several examples of microporous networks based on organic molecules have been shown to be active in various catalytic reactions,7–10 while especially organocatalysis is an emerging topic.1,7,11–16 As microporous polymer networks can be formed by polymerisation of molecular catalysts, no further post-immobilisation process is required. In addition, as the catalytic active sites are a major part of the polymer backbone, the often observed low loading of immobilised molecular catalysts on preformed supports can be avoided. Therefore, due to their high surface area and insoluble nature, these networks combine the benefits of both homogeneous and heterogeneous catalysis. The insolubility enables easy separation after the reaction while the high porosity allows good accessibility of catalytic centres, important to achieve high conversion rates.
Recently we showed that microporous polymer networks based on binaphthyl phosphoric acids can be used in asymmetric heterogeneous organocatalysis.1,7 Using oxidative polymerisation we obtained microporous networks with surface areas up to 386 m2 g−1 that showed very high selectivity in the transfer hydrogenation reaction and C–C bond formation. In this communication we report that the surface area and pore volume of these microporous polymer networks can be tuned by co-polymerisation. This enables us to compare the catalytic performance of the networks in transfer hydrogenations as a function of their surface area and porosity. It is known that co-polymerisation with structure directing motifs of different sizes/lengths can result in networks differing in surface areas and pore volumes.17,18 These porous characteristics are some of the key aspects in catalysis as all reactions take place on interfaces, and therefore an increase in the surface area and pore volume should in principle increase the accessibility of the active centres and thus the reaction rate.
To obtain a catalytically active network we polymerised (R)-3,3′-bis(10-(thiophen-3-yl)anthracen-9-yl)-1,1′-binaphthalen-2,2′-diyl-phosphoric acid (BNPPA) with 1,3,5-tris(2-thienyl)benzene (TTB) in a random oxidative co-polymerisation in monomer ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:5 of BNPPA to TTB (Scheme 1).
:1 and 1:5 of BNPPA to TTB (Scheme 1).
 | ||
| Scheme 1 Monomers used for the co-polymerisation: BNPPA (1) and TTB (2). | ||
BNPPA is known to build porous networks with a surface area of 386 m2 g−1 while networks based on TTB have a surface area of 1060 m2 g−1.19 By mixing these tectons we could obtain networks with increased surface areas compared to the one solely composed of BNPPA (see Table 1). Thus for monomer ratios of 1:1 and 1:5, networks with BET surface areas of 577 m2 g−1 and 668 m2 g−1 and pore volumes of 0.49 cm3 g−1 and 0.58 cm3 g−1 were obtained, respectively. Nitrogen sorption isotherms of the polymer networks are shown in Fig. 1. The ratio of the monomers in the resulting co-polymer networks was determined by ICP-OES and is comparable to the initial ratio. Additionally no iron residues could be detected via this method.
| Polymer network | Surface area/m2 g−1 | Pore volume/cm3 g−1 | TOF/h−1 |
|---|---|---|---|
| TOF was calculated after 65 min reaction time. | |||
| BNPPA network | 386 | 0.28 | 5.24 |
| BNPPA/TTB 1:1 co-polymer |
577 | 0.49 | 7.45 |
| BNPPA/TTB 1:5 co-polymer |
668 | 0.58 | 10.07 |
| TTB network19 | 1060 | 0.71 | — |
| Molecular catalyst | — | — | 12.38 |
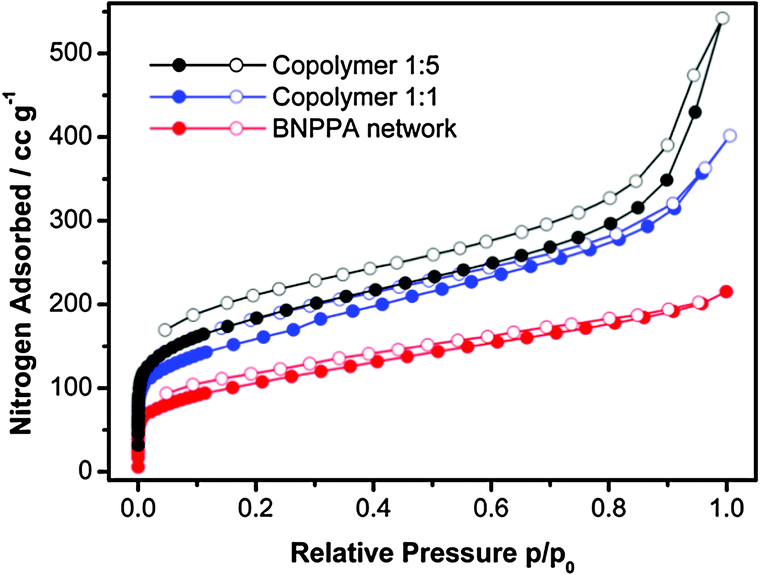 | ||
| Fig. 1 Nitrogen sorption isotherms of polymer networks. | ||
As shown before, the network solely composed of molecule 1 showed excellent performance as an organocatalyst in the transfer hydrogenation on benzoxazines (>99% conv. after 2 h, 98% ee). To study the influence of porosity on the catalytic performance we chose a substrate with lower electron density at which a lower reaction rate can be expected. The kinetic study was therefore performed using 2-phenylquinoline as a substrate which was converted in good yields and selectivity before (99% conv. after 24 h, 99% ee, 5 mol% catalyst).1 The reaction was performed in deuterochloroform at room temperature using 2 mol% of the catalyst only, calculated for the catalytically active centres. We used such little amounts to be able to easily follow the conversion of the heterogeneous reaction by in situ NMR measurements. Blank experiments without any catalyst and with PTTB showed no conversion of the substrate. As the catalyst is covalently bonded in the network, catalyst leaching is improbable as shown in previous studies.1,7 In Fig. 2, the catalytic performance of the pure BNPPA network is compared with the performance of the co-polymers and the molecular catalyst. As expected, the homogeneous catalysis using the molecular catalyst shows a higher conversion under the given conditions. For the network composed of pure BNPPA a conversion of 50% can be obtained after 6 hours. The same reaction was performed with the co-polymer networks using always the same molar amount of the binaphthyl phosphoric acid. Fig. 2 shows the progress of the reaction over time, and the corresponding TOFs are shown in Table 1.
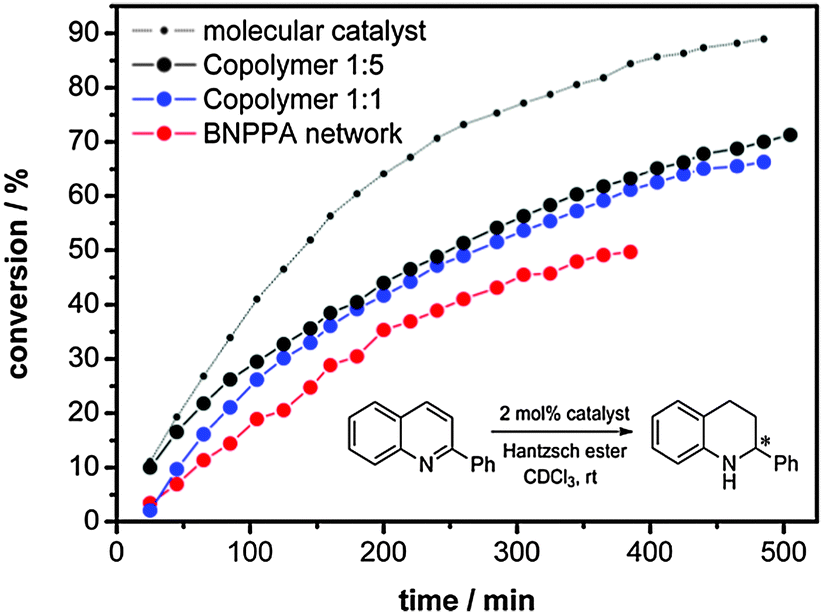 | ||
| Fig. 2 Transfer hydrogenation of 2-phenylquinoline monitored by in situ NMR measurements. | ||
The conversion rate of the co-polymer 1:1 is higher than that obtained for the pure BNPPA catalyst. The reactions with the co-polymer network 1:5 shows even higher conversion than that obtained with co-polymer 1:1, which corresponds to the difference in surface area and porosity.
This experiment shows that the reaction rate can be enhanced by increasing the accessible surface area and pore volume of the microporous polymer network catalysts. This is most probably due to the more easily accessible active centres and the higher diffusion rate of substrates within the network which enables the possibility to tune porosity in a way to fit to particular substrates and their reaction conditions. It should be however noted that porous characteristics are measured at low pressures at 77 K, and thus are actually not comparable to the situation in a reaction mixture, i.e. in solvents at elevated temperatures. For example, some swelling of the polymer networks in the reaction mixture has to be assumed, as this has been even observed to happen during nitrogen sorption measurements.20–23 This swelling should increase the accessibility of catalytic centres, and even materials which showed lower surface areas in nitrogen sorption measurements can be quite active catalysts under realistic reaction conditions. However, the measurements presented here prove that there is an influence of the intrinsic porosity of microporous polymers on the catalytic performance. Thus the accessibility of active sites is provided not only by swelling of polymer networks, but also by their open channel structure.
In summary, we synthesised microporous polymer networks based on BNPPA that can be used to bridge the gap between asymmetric homogeneous and heterogeneous catalysis. The control over the surface area and pore volume of the polymer networks via co-polymerisation enables the possibility to study their influence on the catalytic performance. We could show that polymer networks with higher surface areas show higher reaction rates in asymmetric transfer hydrogenation. This indicates that the catalytic reaction takes place inside the network and the porosity directly influences the diffusion and accessibility of the active catalytic centres.
We acknowledge financial support from the European Research Council within the project 278593_ORGZEO and the Deutsche Forschungsgemeinschaft (DFG) within the cluster of excellence “UNICAT – Unifying Concepts in Catalysis”.
Notes and references
- D. S. Kundu, J. Schmidt, C. Bleschke, A. Thomas and S. Blechert, Angew. Chem., Int. Ed., 2012, 51, 5456–5459 CrossRef CAS PubMed.
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS PubMed.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- Y. G. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS PubMed.
- Q. Sun, Y. Jin, L. Zhu, L. Wang, X. Meng and F.-S. Xiao, Nano Today, 2013, 8, 342–350 CrossRef CAS PubMed.
- C. Bleschke, J. Schmidt, D. S. Kundu, S. Blechert and A. Thomas, Adv. Synth. Catal., 2011, 353, 3101–3106 CrossRef CAS.
- Z. G. Xie, C. Wang, K. E. deKrafft and W. B. Lin, J. Am. Chem. Soc., 2011, 133, 2056–2059 CrossRef CAS PubMed.
- J. X. Jiang, C. Wang, A. Laybourn, T. Hasell, R. Clowes, Y. Z. Khimyak, J. L. Xiao, S. J. Higgins, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2011, 50, 1072–1075 CrossRef CAS PubMed.
- X. Du, Y. L. Sun, B. E. Tan, Q. F. Teng, X. J. Yao, C. Y. Su and W. Wang, Chem. Commun., 2010, 46, 970–972 RSC.
- Y. Zhang, Y. Zhang, Y. L. Sun, X. Du, J. Y. Shi, W. D. Wang and W. Wang, Chem.–Eur. J., 2012, 18, 6328–6334 CrossRef CAS PubMed.
- M. Rose, A. Notzon, M. Heitbaum, G. Nickerl, S. Paasch, E. Brunner, F. Glorius and S. Kaskel, Chem. Commun., 2011, 47, 4814 RSC.
- K. Thiel, R. Zehbe, J. Roeser, P. Strauch, S. Enthaler and A. Thomas, Polym. Chem., 2013, 4, 1848–1856 RSC.
- J. Roeser, K. Kailasam and A. Thomas, ChemSusChem, 2012, 5, 1793–1799 CrossRef CAS PubMed.
- C. A. Wang, Z. K. Zhang, T. Yue, Y. L. Sun, L. Wang, W. D. Wang, Y. Zhang, C. Liu and W. Wang, Chem.–Eur. J., 2012, 18, 6718–6723 CrossRef CAS PubMed.
- E. Merino, E. Verde-Sesto, E. M. Maya, M. Iglesias, F. Sanchez and A. Corma, Chem. Mater., 2013, 25, 981–988 CrossRef CAS.
- J. Schmidt, M. Werner and A. Thomas, Macromolecules, 2009, 42, 4426–4429 CrossRef CAS.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS PubMed.
- J. Schmidt, J. Weber, J. D. Epping, M. Antonietti and A. Thomas, Adv. Mater., 2009, 21, 702–705 CrossRef CAS.
- D. Z. Tan, W. J. Fan, W. N. Xiong, H. X. Sun, Y. Q. Cheng, X. Y. Liu, C. G. Meng, A. Li and W. Q. Deng, Macromol. Chem. Phys., 2012, 213, 1435–1440 CrossRef CAS.
- J. Weber, J. Schmidt, A. Thomas and W. Bohlmann, Langmuir, 2010, 26, 15650–15656 CrossRef CAS PubMed.
- J. Weber, M. Antonietti and A. Thomas, Macromolecules, 2008, 41, 2880–2885 CrossRef CAS.
- J. X. Jiang, A. Trewin, F. B. Su, C. D. Wood, H. J. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, Macromolecules, 2009, 42, 2658–2666 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, synthetic details, and NMR spectra. See DOI: 10.1039/c3cc49220f |
| This journal is © The Royal Society of Chemistry 2014 |