 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of pharmaceutically relevant 17-α-amino steroids using an ω-transaminase†
Nina
Richter
ab,
Robert C.
Simon
c,
Wolfgang
Kroutil
bc,
John M.
Ward
d and
Helen C.
Hailes
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: h.c.hailes@ucl.ac.uk; Tel: +44 (0)20 7679 4564
bACIB GmbH, c/o Department of Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria
cDepartment of Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria
dDepartment of Biochemical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK
First published on 19th February 2014
Abstract
An efficient and sustainable biocatalytic route for the synthesis of important 17-α-amino steroids has been developed using an ω-transaminase variant from Arthrobacter sp. Optimisation of the reaction conditions facilitated the synthesis of these valuable synthons on a preparative scale, affording excellent isolated yields and stereocontrol.
Steroids are a large and diverse class of secondary metabolites, essential for the control of a variety of biological processes. Based on this key role in metabolism, steroids and their derivatives often exhibit biological activity and therefore have enormous potential as pharmaceuticals.1 Indeed, approximately 300 steroidal drugs have been placed on the market since 1950 with cortisone as one prominent example.1,2 Moreover, among the 200 top-selling drugs in 2010 13% were steroids and derivatives thereof.3 17-Amino steroids (1) (Fig. 1) have proven to be particularly interesting non-natural steroids that are used as intermediates in the synthesis of biologically active steroidal derivatives. For example the 17β-arylsulfonamide derivative 2 has recently been highlighted as a potent inhibitor of steroid sulfatase, a target in the treatment of breast cancer.4–7 Moreover, a number of 17β-aminoestrogens were identified to have a prolonged anticoagulant effect in rodents.8 In both examples the amine functionality, and derivatives thereof, attached to the C-17 position of the steroid is presumed to be crucial for the biological activity.
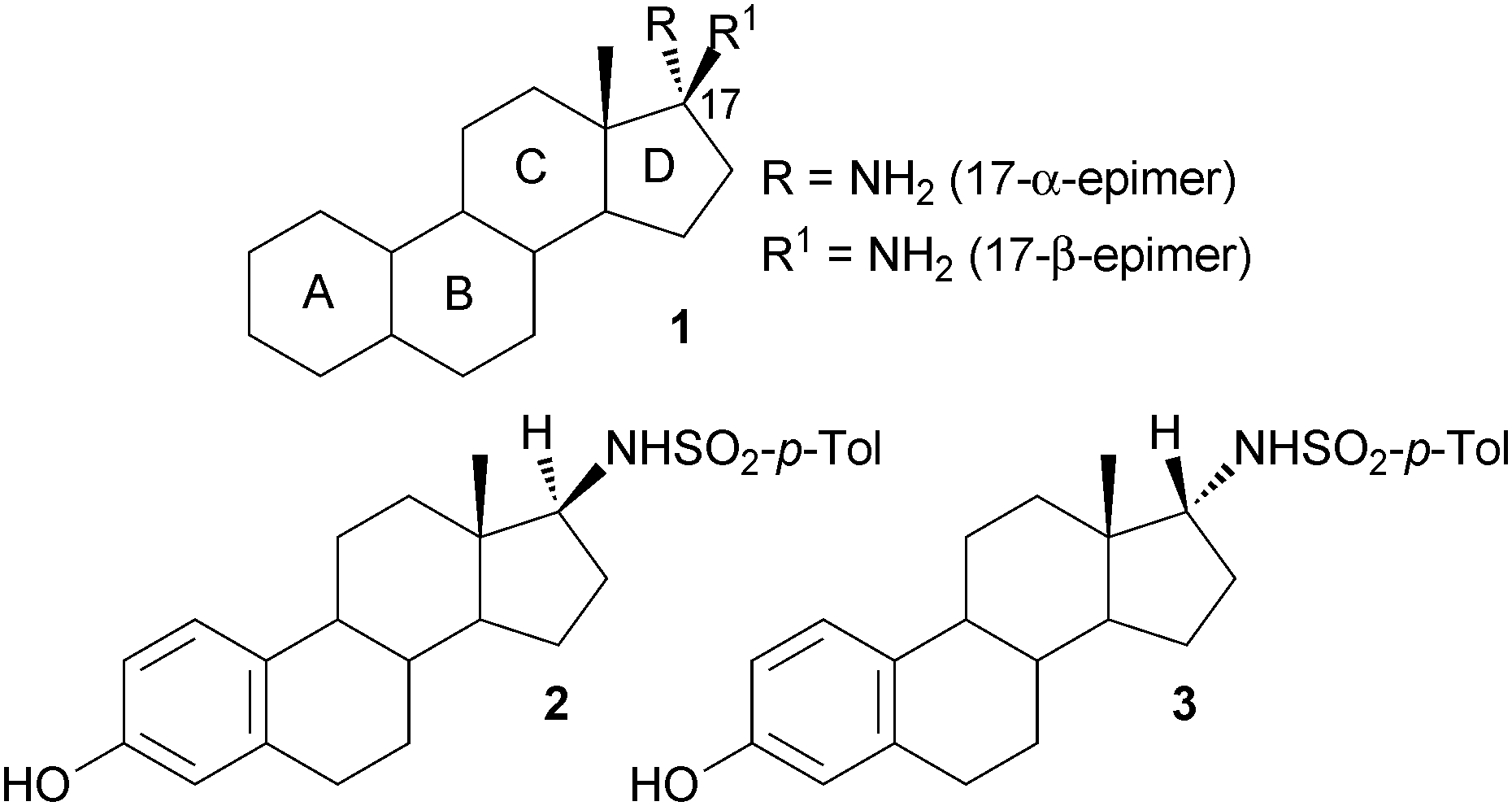 | ||
| Fig. 1 Steroid core structure 1 with amino group at C-17, and 17β- and 17α-sulfonamide derivatives 2 and 3, respectively. | ||
In general, the 17β-amino steroid motif is accessible by a classical two-step method, via reductive amination and deprotection, and consequently studies have focussed on derivatives of the β-epimer.6,7 Although the synthesis of the 17α-amino steroid is less efficient, requiring a three-step synthetic route with a low yielding reduction reaction, the α-epimer derivative 3 (Fig. 1) was also found to be a potent sulfatase inhibitor.6,8 As a consequence of the poor accessibility of the α-epimer the potential of further derivatives has to the best of our knowledge not been investigated to date. Therefore, the development of an efficient method for the synthesis of the α-epimer would be highly desirable. Here we present a novel route to access 17-α-amino steroids by applying the use of an ω-transaminase (ω-TAm). ω-TAms are continuing to attract significant attention for use in asymmetric synthesis for the generation of both (S)- and (R)-chiral amines.9–19 Moreover, biocatalytic strategies employing ω-TAms have been used for the synthesis of pharmaceutically relevant compounds,20–27 even leading to the development of an industrial process.28
In initial studies our aim was to use two previously reported ω-TAms, Vibrio fluvialis (Vf-TAm),10 and Chromobacterium violaceum DSM30191 (CV-TAm),16 and a particularly interesting ω-TAm variant described, from Arthrobacter sp. (ArRMut11).28 CV-TAm has previously been reported for the transamination of a wide range of ketones including more sterically challenging substrates such as 1,3-dihydroxy-1-phenylpropan-2-one,16,25 while Vf-TAm has been used with in general smaller substrates.11,14 The ω-TAm ArRMut11 variant was evolved by Savile and co-workers over 11 rounds of mutation, to catalyse the amination of sterically demanding 1,3-ketoamides to generate the (R)-aminoamide functionality present in sitagliptin.28 An additional feature of this variant is its tolerance towards high co-solvent concentrations and 2-propylamine, facilitating the use of this low cost amine donor to shift the equilibrium towards the product.11,17,28–31 The variant ArRMut11 has also been successfully used for the transamination of tetralone and chromone bicyclic compounds.29,30 For the generation of 17α-amino steroids TAms are required with good co-solvent tolerance, due to the limited aqueous solubilities of the substrates, and ability to accept the large tetracyclic steroidal ring system. Here we describe the use of the three ω-TAms and optimisation of the reaction conditions using ArRMut11 as a novel route to 17-α-amino steroids.
To evaluate the potential of the selected ω-TAms in the asymmetric amination of steroid precursors, the truncated analogue 4 and steroid 5 (10 mM) were initially screened against the three ω-TAms using either (R) or (S)-α-methylbenzylamine as the amine donor, depending on the selectivity of the ω-TAm used, and acetophenone production was monitored by HPLC analysis.
Only for ArRMut11 were conversions >5% detected, so this ω-TAm was used in further experiments with 4–8 (10 mM) using isopropylamine as a low cost donor (Scheme 1) and 20% of either 1,2-dimethoxyethane (DME) (4 and 5) or dimethylsulfoxide (DMSO) (6–8) as co-solvent. LC-MS analysis indicated that 5–7 were good substrates for the ω-TAm. Although substrates 4 and 8 seemed to be accepted by the enzyme and m/z peaks corresponding to the product were detected by LC-MS, it was not possible to isolate a single product. This was not surprising due to the two ketone moieties in 4 complicating product analysis, and α,β-unsaturated enones in both 4 and 8 which can form conjugated enamines in the presence of amines. Compounds 4 and 8 were therefore not explored further.
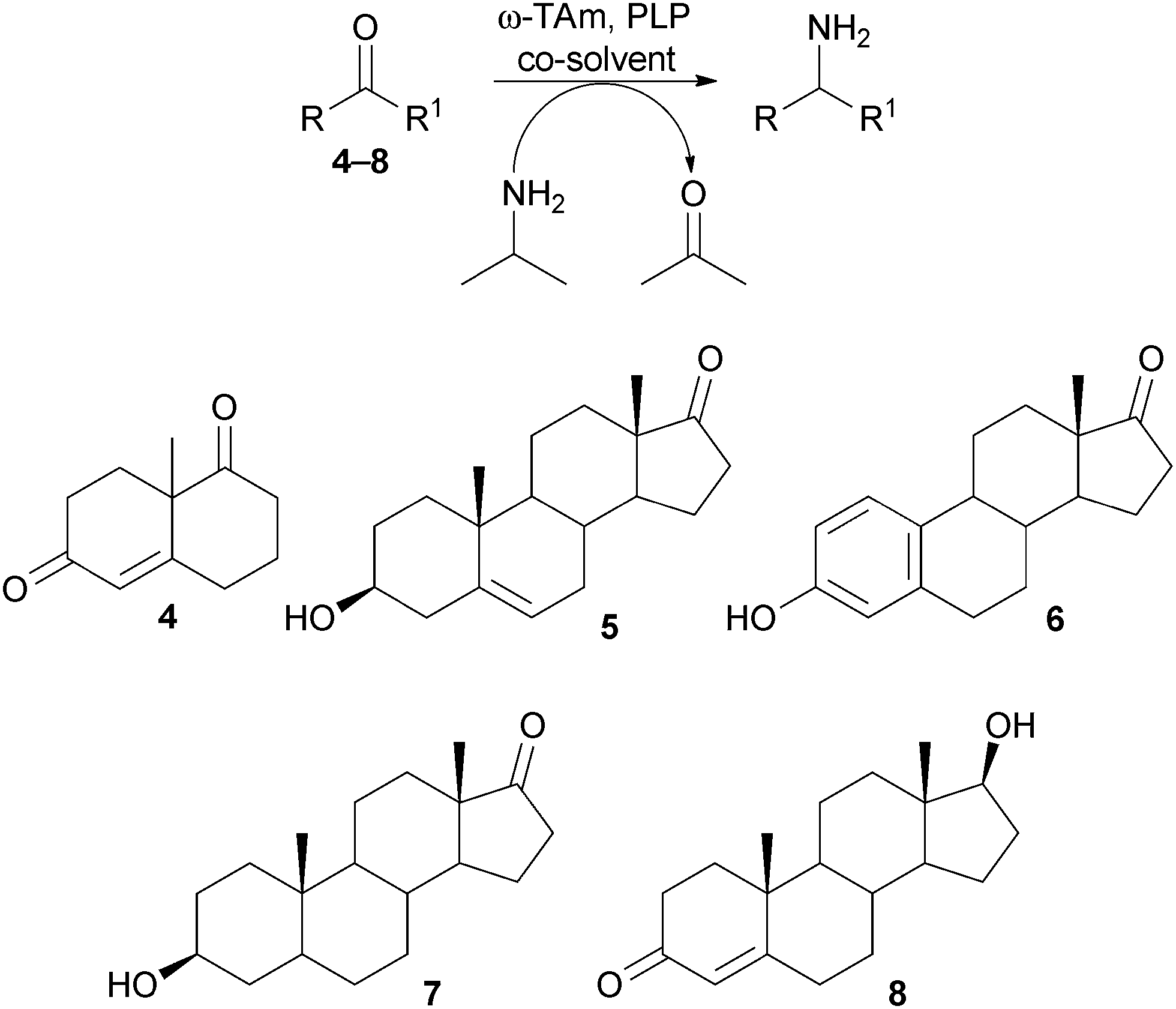 | ||
| Scheme 1 TAm reaction using ArRMut11 and substrates 4–8. | ||
The reaction conditions were then optimised in order to maximise reaction rates and conversion yields using trans-dehydroandrosterone (5) as a model substrate (1 mL, 10 mM). Parameters including the employment of co-solvents, to increase the substrate solubility, as well as the amount of amine donor 2-propylamine, to maximise the conversion rate were explored. DMSO is frequently used as a co-solvent in biocatalysis, but the substrates were not fully soluble in mixtures of DMSO–water. DME and dimethylformamide (DMF) have recently been applied successfully as co-solvents in ω-TAm reactions so their potential here was investigated.32 Both DME and DMF enhanced the solubility of the substrates in mixed aqueous systems giving rise to clear rather than cloudy solutions, whereas substrates exhibited the best solubilities in water–DMF mixtures. Comparative studies using 25% of DME or DMF as co-solvents led to similar conversions (∼60%) after a reaction time of 6 days (Fig. S1a, ESI†), however since higher reaction rates were observed with DMF it was selected as a co-solvent for all further experiments. The impact of different DMF concentrations (25–50%) on reaction rate and conversion were investigated as well. While conversions after 6 days were again rather similar (64–68%) the fastest reaction rate and highest conversion (68%) were reached using 35% of DMF as an optimal co-solvent concentration.
The concentration of the amine donor 2-propylamine was then investigated using 20-, 50- and 100-fold molar excesses to shift the equilibrium of the ω-TAm reaction towards the aminated steroid. The conversion of 5 to 17-amino-3β-hydroxyandrost-5-ene was improved from 40% after 6 days using a 20-fold excess of 2-propylamine to 81% after 5 days using a 100-fold excess (Fig. S1b, ESI†). The reaction profile for the biotransformation using the optimised conditions (35% DMF and 100-fold molar excess of 2-propylamine) over a period of 6 days is shown in Fig. 2. The data indicated that the reaction conditions were well tolerated by ω-TAm ArRMut11.
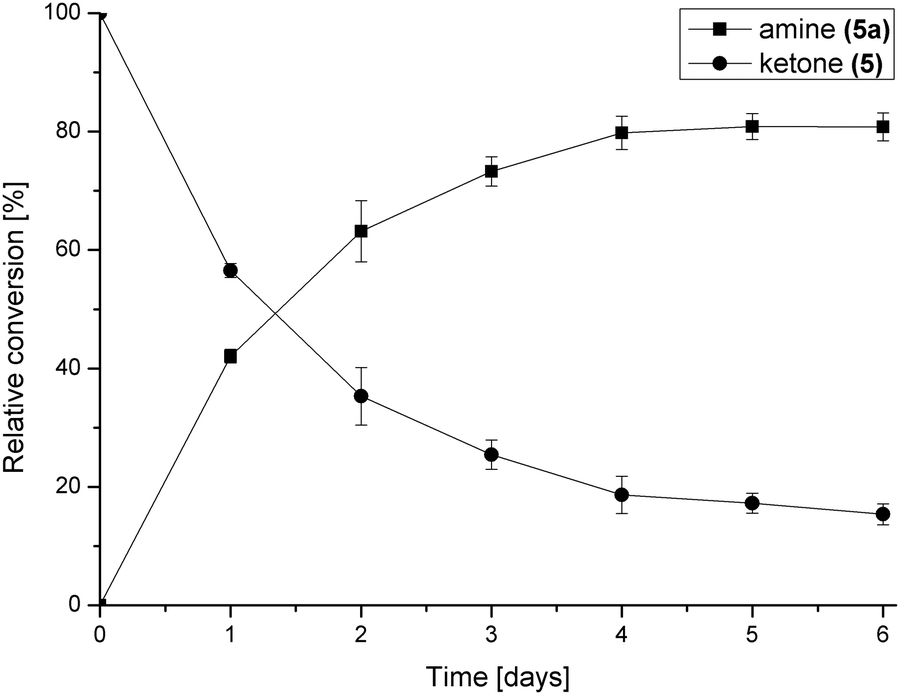 | ||
| Fig. 2 Reaction profile of the asymmetric amination of 5 (1 mL, 10 mM) using the optimised conditions: ω-TAm ArRMut11, 35% v/v DMF–water and 100-fold molar excess of 2-propylamine (1 M). | ||
As a consequence these optimised conditions were used for the asymmetric amination of steroids 6 and 7 (1 mL, 10 mM). These were successfully transformed to the corresponding 17-amino steroids with excellent conversions, yielding 17-amino-1,3,5(10)-estratrien-3-ol from 6 in 68% yield and 17-amino-5α-androstan-3β-ol from 7 in 71% yield (Table S1, ESI†). The transamination of 5–7 was then performed on preparative scale (50 mg substrate; 20 mL, 10 mM). Reactions were stopped after three days and the conversion and stereoselectivity of the reaction determined by GC and NMR analysis. Interestingly, on a preparative scale all biotransformations showed enhanced conversions compared to the previous results despite a shorter reaction time (Table 1cf. Table S1, ESI†): however for the synthesis of 5a this could be ascribed to the slightly higher amounts of enzyme used. In particular, the amination of steroid 6 was improved 1.4 fold, giving amine 6a with a conversion of 96%. Amines 5a and 7a were also produced with excellent conversions of 88% and 90%, respectively. To establish the stereoselectivities reaction products were purified by flash silica chromatography, and isolated in yields of 83% (5a), 85% (6a) and 89% (7a).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
:
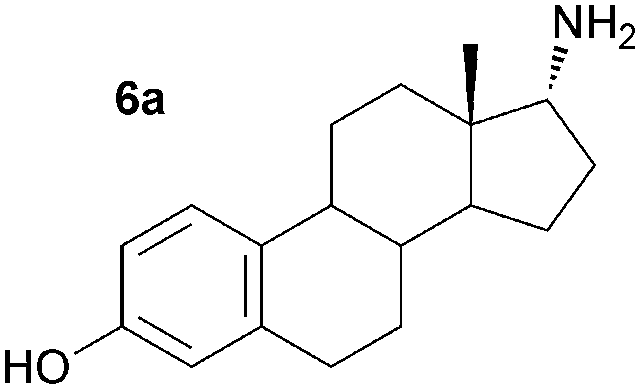
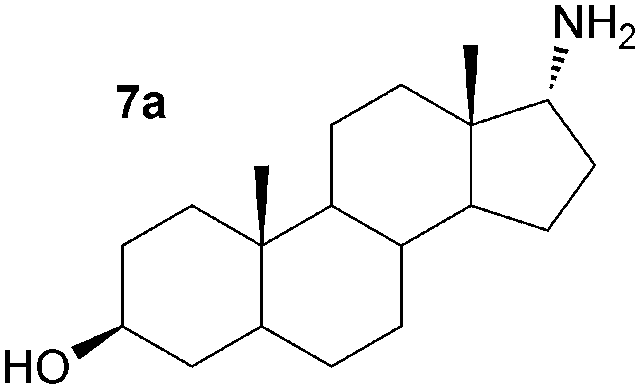
Assignment of the absolute configuration of the products was carried out by comparing the previously reported NMR spectroscopic data to that of 6a and using 1H NMR NOESY experiments (between 17-H and the methyl group at C-13).8 These indicated that the 17-α-epimer (anti) was formed with full stereocontrol in all three cases. The newly formed chiral amine moiety has an (R)-configuration, in agreement with previous stereoselectivities reported using the (R)-selective ω-TAm ArRMut11. This biocatalytic approach gives the 17-α-amino steroids in one step, starting from the corresponding carbonyl compound, in excellent isolated yields of 83–89%, and highlights the tolerance of the ω-TAm ArRMut11 to such sterically challenging substrates. Compared to the classical chemical synthesis of 6a reported not only was the number of synthetic steps reduced from three to one, but also the overall yield was improved 9-fold.8
In summary, a highly stereoselective, efficient and sustainable biocatalytic route, facilitating access to a variety of highly desirable 17-α-amino steroids has been developed. After optimisation of the reaction parameters the 17-α-amino steroids were synthesised in high isolated yields of 83–89% via a one-step procedure on a preparative scale. This novel biocatalytic methodology enables access to the α-epimer of key intermediates in the synthesis of biologically active steroidal derivatives. Moreover, the described method represents the shortest routes towards 17-α-amino steroids published to date.
This work has been supported by a postdoctoral fellowship of the German academic exchange service (DAAD) to N. R.; part of the work was supported by the Austrian BMWFJ, BMVIT, SFG, Standortagentur Tirol and ZIT through the Austrian FFG-COMET-Funding Program.
Notes and references
- C. H. L. Shackleton, Lipids, 2012, 47, 1–12 CrossRef CAS PubMed.
- A. S. Chapelon, D. Moraleda, R. Rodriguez, C. Ollivier and M. Santelli, Tetrahedron, 2007, 63, 11511–11616 CrossRef CAS PubMed.
- D. J. Mack, M. L. Weinrich, E. Vitaku and J. T. Njardarson, Top 200 Brand Name Drugs by US Retail Sales in 2010, http://cbc.arizona.edu/njardarson/group/top-pharmaceuticals-poster.
- J. Geisler, H. Sasano, S. A. Chen and A. Purohit, J. Steroid Biochem. Mol. Biol., 2011, 125, 39–45 CrossRef CAS PubMed.
- R. Maltais and D. Poirier, Steroids, 2011, 76, 929–948 CrossRef CAS PubMed.
- Y. A. Mostafa and S. D. Taylor, Bioorg. Med. Chem., 2012, 20, 1535–1544 CrossRef CAS PubMed.
- S. D. Taylor and J. Harris, Steroids, 2011, 76, 1098–1102 CrossRef CAS PubMed.
- C. Lemini, E. Cruz-Ramos, R. A. Toscano and R. Cruz-Almanza, Steroids, 1998, 63, 556–564 CrossRef CAS.
- J. Ward and R. Wohlgemuth, Curr. Org. Chem., 2010, 14, 1914–1927 CrossRef CAS.
- J. S. Shin, H. Yun, J. W. Jang, I. Park and B. G. Kim, Appl. Microbiol. Biotechnol., 2003, 61, 463–471 CrossRef CAS PubMed.
- M. S. Malik, E. S. Park and J. S. Shin, Appl. Microbiol. Biotechnol., 2012, 94, 1163–1171 CrossRef CAS PubMed.
- G. Shin, S. Mathew, M. Shon, B. G. Kim and H. Yun, Chem. Commun., 2013, 49, 8629–8631 RSC.
- M. Höhne and U. T. Bornscheuer, ChemCatChem, 2009, 1, 42–51 CrossRef.
- S. Mathew and H. Yun, ACS Catal., 2012, 2, 993–1001 CrossRef CAS.
- B. Wang, H. Land and P. Berglund, Chem. Commun., 2013, 49, 161–163 RSC.
- U. Kaulmann, K. Smithies, M. E. B. Smith, H. C. Hailes and J. M. Ward, Enzyme Microb. Technol., 2007, 41, 628–637 CrossRef CAS PubMed.
- K. E. Cassimjee, C. Branneby, V. Abedi, A. Wells and P. Berglund, Chem. Commun., 2010, 46, 5569–5571 RSC.
- M. D. Truppo, J. D. Rozzell and N. J. Turner, Org. Process Res. Dev., 2010, 14, 234–237 CrossRef CAS.
- M. D. Truppo, J. D. Rozzell and N. J. Turner, Org. Biomol. Chem., 2010, 8, 1280–1283 CAS.
- E. S. Park, J. Y. Dong and J. S. Shin, ChemCatChem, 2013, 12, 3538–3542 CrossRef.
- I. K. Mangion, B. D. Sherry, J. J. Yin and F. J. Fleitz, Org. Lett., 2012, 14, 3458–3461 CrossRef CAS PubMed.
- M. Fuchs, D. Koszelewski, K. Tauber, W. Kroutil and K. Faber, Chem. Commun., 2010, 46, 5500–5502 RSC.
- E. Siirola, F. G. Mutti, B. Grischek, S. F. Hoefler, W. M. F. Fabian, G. Grogan and W. Kroutil, Adv. Synth. Catal., 2013, 355, 1703–1708 CrossRef CAS.
- M. E. B. Smith, B. H. Chen, E. G. Hibbert, U. Kaulmann, K. Smithies, J. L. Galman, F. Baganz, P. A. Dalby, H. C. Hailes, G. J. Lye, J. M. Ward, J. M. Woodley and M. Micheletti, Org. Process Res. Dev., 2010, 14, 99–107 CrossRef CAS.
- K. Smithies, M. E. B. Smith, U. Kaulmann, J. L. Galman, J. M. Ward and H. C. Hailes, Tetrahedron: Asymmetry, 2009, 20, 570–574 CrossRef CAS PubMed.
- C. Molinaro, P. G. Bulger, E. E. Lee, B. Kosjek, S. Lau, D. Gauvreau, M. E. Howard, D. J. Wallace and P. D. O'Shea, J. Org. Chem., 2012, 77, 2299–2309 CrossRef CAS PubMed.
- T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772–6775 CrossRef CAS PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, J. H. Sattler and W. Kroutil, Adv. Synth. Catal., 2011, 353, 3227–3233 CrossRef CAS.
- D. Pressnitz, C. S. Fuchs, J. H. Sattler, T. Knaus, P. Macheroux, F. G. Mutti and W. Kroutil, ACS Catal., 2013, 3, 555–559 CrossRef CAS.
- E. S. Park, J. Y. Dong and J. S. Shin, Org. Biomol. Chem., 2013, 11, 6929–6933 CAS.
- R. C. Simon, C. S. Fuchs, H. Lechner, F. Zepeck and W. Kroutil, Eur. J. Org. Chem., 2013, 3397–3402 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures and characterisation data for 5a, 6a, 7a. Supplementary Fig. S1, Table S1 and NMR data for 5a, 6a, 7a. See DOI: 10.1039/c3cc49080g |
| This journal is © The Royal Society of Chemistry 2014 |