Catalyst-mediated yet catalyst-free hydrogels formed by interfacial chemical activation†
Eunkyoung
Byun‡
a,
Ji Hyun
Ryu‡
b and
Haeshin
Lee
*ab
aDepartment of Chemistry, KAIST Institute NanoCentury (CNiT), Korea Advanced Institute of Science and Technology (KAIST), Daejeon 305-701, Republic of Korea. E-mail: haeshin@kaist.ac.kr
bGraduate School of Nanoscience and Technology, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 305-701, Republic of Korea
First published on 20th January 2014
Abstract
We introduce an entirely new method for forming hydrogels, wherein the formation is triggered by a catalyst, but the resulting hydrogels are catalyst-free. We unexpectedly found that a sufficient amount of crosslinkable catecholquinone/semiquinone free radicals is generated by the physical contact between catechol-containing polymers and surface-immobilized catalysts.
Hydrogels, swollen matrices of crosslinked structures in biological fluids, have attracted much attention as tissue engineering scaffolds, drug delivery depots, macromolecule biosensors, and tissue adhesives in biomedical fields.1 Important aspects to be considered during the preparation of hydrogels for biomedical applications are controllable crosslinking, mechanical properties, and excellent biocompatibility. Controllable in situ hydrogels have often utilized covalent crosslinking chemistry using biological or chemical catalysts. The covalent bonds generated by biological catalysts such as transglutaminase, tyrosinase, or horseradish peroxidase to fabricate hydrogels are representative examples.2 To demonstrate the safety of biomedical materials intended for human use, appropriate tests related to the stability are required for biological evaluation, including in vitro cytotoxicity, sensitization, hemocompatibility, genotoxicity, carcinogenicity, reproductive and developmental toxicity, and biodegradation investigations. However, one of the significant limitations in human clinical use is the unavoidable incorporation of biological catalysts. This might cause a possibility of unexpected toxicity in the human body, which includes a series of toxicological evaluation.3 Thus, the development of a strategy for obtaining a catalyst-mediated hydrogel that does not contain the utilized biological catalysts within the hydrogels is ideal. Even in the case of hydrogels produced by chemical catalysts such as copper bromide, copper iodide, or copper sulfate, as used in click chemistry, this important issue cannot be avoided.4
We hypothesized that catalysts could be completely removed from hydrogels by the following strategy. First, catalysts should be immobilized onto solid substrates. Second, the product resulting from the catalyst reaction should not be the final product. Rather, the activated product should be a chemical intermediate that can react with the counterparts at a later time to form the final crosslinked product. Furthermore, the intermediate should be stable under physiological conditions. Simple immobilization of the existing catalysts on a surface cannot achieve catalyst-excluded hydrogels. For example, the crosslinking reaction between a free amine group and the acyl group at the side chain of glutamine occurs only at the catalytic site of transglutaminase. Thus, enzyme immobilization typically results in local crosslinking reactions at the interface between the enzyme and the polymer, but crosslinking does not occur in the bulk polymer solution. In the case of chemical catalysts such as copper(I), for click chemistry, the indispensible catalyst immobilization step is even more difficult than that of enzymes.
Hematin is an iron-containing hydroxyferri-porphyrin that catalyzes the oxidation of phenol- or catechol-containing molecules in the presence of H2O2. The intermediates generated by the catalytic activity of hematin, the so-called quinone/semiquinone free radical (SFR), have been reported to be relatively stable under aqueous conditions.5 The established stable quinone intermediate can later react with amine-containing or catechol-containing molecules to form C–C/C–O/C–N couplings in aromatic rings.6 Therefore, the utilization of hematin and catechol-containing polymers presents a good model system for testing our hypothesis. We previously reported hydrogel formation with a water-soluble, hematin–chitosan conjugate.7 However, similar to previous reports on other catalyst-mediated hydrogels, excluding the hematin catalyst from the hydrogel was not possible. It is noted that hematin binds and disrupts cell membranes that causes the cytotoxicity of cells at a high concentration.8 Thus, developing a general strategy in which the hydrogel is formed by catalysts but the resulting hydrogel does not contain the catalysts remains a great challenge.
In this report, we introduce a method for fabricating a catalyst-free hydrogel for the first time. Hematin was chosen as a model catalyst, and catechol-containing polymers (e.g., poly(ethylene glycol) and a chitosan; structures shown in Fig. S1b and c, ESI†) were used as hydrogel materials. To achieve catalyst-free hydrogels, catalysts (i.e., hematin) were immobilized on the inner surface of a poly(propylene) syringe. This step satisfies the first necessary condition described above. The quinone/SFR species, generated by catechol oxidation via the catalytic activity of hematin, were created when the polymer solution passed through the syringe. The quinone/SFR species were not the final products of the hematin catalytic reaction, but chemically active intermediates. This process satisfies the second necessary condition. We found that the flow-through solution was in a sol state for the first 15 min, but soon after, the solution changed to a gel state with an elastic modulus of 104 Pa. The crosslinking chemistry responsible for this change arises due to the presence of catechol–catechol and catechol–amine couplings. Inductively coupled plasma-optical emission spectroscopy (ICP-OES) and electron paramagnetic resonance (EPR) analysis demonstrated that hematin was not present in the hydrogels. This new hydrogel formation method is advantageous over previous catalyst-mediated hydrogels in that the strict toxicity concern due to the incorporated catalyst in hydrogels is reduced, and the widely used double-channel syringe is no longer needed. Compared with the physically crosslinked hydrogels driven by electrostatic or hydrophobic interactions,9 which can be categorized as catalyst-free gels, the one described herein is expected to control the mechanical strength as well as the in vivo and in vitro stability. Also, the hydrogels formed by electrostatic interactions are also catalyst-free. However, these hydrogels are not suitable because of the rapid gelation kinetics. Thus, the immediate gelation behavior is not compatible with a variety of medical devices such as catheters and endoscopes whose configuration shows a long, single tubing.9
A detailed illustration of catalyst-free hydrogel formation is shown in Scheme 1. We prepared the chitosan–catechol conjugate with a degree of catechol conjugation of approximately 4.8%, as determined by ultraviolet–visible (UV-Vis) spectroscopy. The details of the method are described by K. Kim et al.10 Briefly, 3,4-dihydroxyhydrocinnamic acid (HCA) was chemically activated and subsequently reacted with the amine groups of the chitosan. Thus, in principle, when the hematin-immobilized syringe (left bottom gray box, Scheme 1) contains the chitosan–catechol polymers, the catechol moieties are converted to quinone/SFR by the catalytic activity of hematin. The amount of quinone/SFR increases with a reciprocating action (i.e., mixing effect) of a piston (t0–t2). Unexpectedly, just three to four cycles of the reciprocating action of the piston are sufficient to generate the necessary amount of intermediates. Finally, the solution is ejected from the syringe and undergoes a phase transition from the sol to gel state (t3).
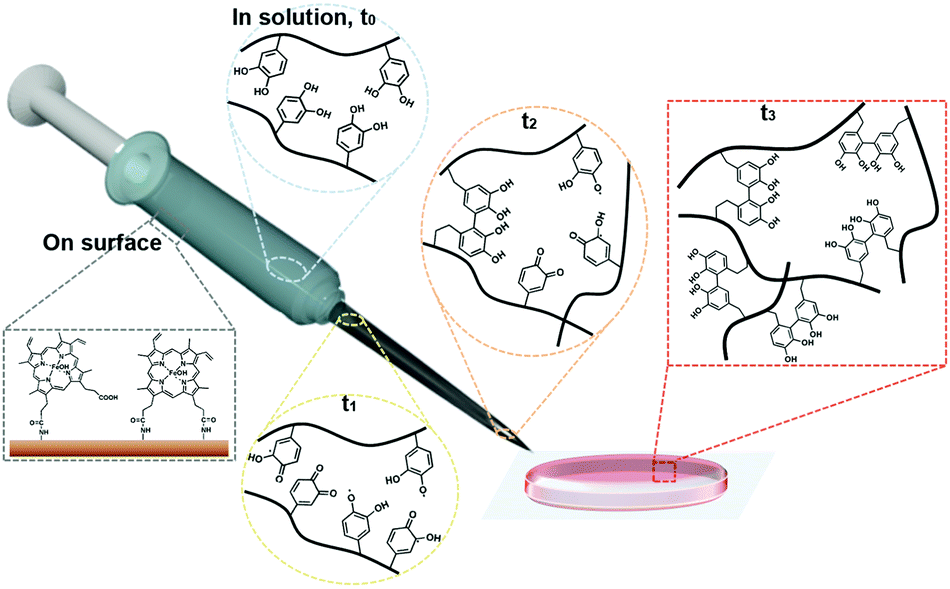 | ||
| Scheme 1 Schematic diagram of the preparation of catalyst-free injectable hydrogels produced by the interfacial chemical activation of surface-immobilized hematin. Via an interfacial reaction between the catechol-containing polymers (t0) and the immobilized hematin in the syringe (shown in gray), the tethered catechol moieties become active intermediates (t1–t2). Finally, the intermediates react with each other, forming catalyst-free hydrogels (t3). | ||
The preparation of a hematin-immobilized surface is critical for achieving a catalyst-free hydrogel. One important consideration is that syringes are typically produced from synthetic polymers such as poly(propylene), making it difficult to functionalize the surface. Thus, we applied the poly(dopamine) (pDA) coating approach (structure shown in Fig. S1a, ESI†).11 Using this method, we can introduce surface amine groups with ease. The pDA coating step is preceded by the application of alkaline conditions overnight. The surface primary amines react with the carboxyl group of hematin through EDC/NHS coupling to immobilize the hematin (Fig. 1a). To confirm the immobilization of hematin on the surface, UV-Vis spectroscopy was used to determine the existence of hematin on the surface, showing prominent absorption peaks at 400 nm and near 600 nm, which indicates the presence of hematin (red, Fig. 1b).12 In addition, X-ray photoelectron spectroscopy (XPS) verified the existence of iron in hematin on the surface. Compared to poly(propylene) syringes and pDA-coated syringes, the hematin-immobilized syringes exhibited Fe2p peaks at 710.9 eV (3/2) and 724.5 eV (1/2) (red, Fig. 1c). In contrast, the Fe2p photoelectron was not detected in the pDA coating (blue) and the unmodified poly(propylene) surface (black). These results indicate that hematin was certainly immobilized by EDC/NHS coupling after the introduction of a pDA layer on the polypropylene syringes.
 | ||
| Fig. 1 (a) Schematic representation of the overall process of hematin immobilization on the syringe surface: after pDA coating (brown) and hematin immobilization by EDC/NHS coupling (green). (b) UV-Vis spectra of the unmodified surface (black), the pDA-coated surface (blue), and the hematin-immobilized surface (red). (c) XPS data (high-resolution Fe2p1/2 and Fe2p3/2) of unmodified (black), pDA-coated (blue), and hematin-immobilized surfaces (red). (d) EPR spectra of the 4-arm-PEG-catechol before (black) and after (red) hematin exposure. (e) UV-Vis absorbance at 330 nm, to monitor the oxidation of the 4-arm-PEG-catechol at 0, 30 min, 1.5 h, 6 h, and 12 h after 3 min of hematin activation. | ||
To confirm the generation of the quinone/SFR in bulk solution by interfacial contact with the hematin-immobilized surfaces, we performed EPR spectroscopy. A four-armed poly(ethylene) glycol-catechol (4-arm-PEG-catechol) was prepared by the conjugation of a catechol to 4-arm-PEG-NH2. To demonstrate the presence of the radical in bulk solution, initiated by hematin activity, the 4-arm-PEG-catechol (0.2 wt%) was dissolved in distilled and deionized water and mixed with 100 mM H2O2. The mixture solution was exposed to hematin-immobilized surfaces for 3 min and then collected for EPR analysis. Fig. 1d shows the EPR spectrum of the 4-arm-PEG-catechol. Before hematin activation, examination by EPR spectroscopy did not show any noticeable signals (black, Fig. 1d). Direct contact of the 4-arm-PEG-catechol with the immobilized hematin caused oxidation of the catechols, converting them to quinone radicals, which were detected by EPR spectroscopy (red, Fig. 1d).13 The detection of organic radicals is important in that the amount of radicals in the bulk polymer solution is large enough for the subsequent covalent crosslinking reaction to produce catechol–catechol crosslinking. It was unexpected that the hydrogel formation would occur via chemical activation by the immobilized hematin with such a short exposure time (approximately 3 s). Importantly, the EPR detection indicates that the organic quinone radicals are stable for the subsequent inter-molecular catechol–catechol reaction. The key issue in this study is whether the chemically activated polymers can react with each other after injection in the absence of the catalysts. Thus, we monitored UV absorption changes in the solution at 330 nm, which verified catechol–catechol crosslinking between the 4-arm-PEG-catechol chains after injection (Fig. 1e). As expected, the catechol–catechol reaction occurred in the absence of the catalyst; the collected polymer solutions exhibited an increase in A330 as a function of time (black dot), indicating a subsequent crosslinking reaction. After a 3 min exposure to the immobilized hematin, the polymer solution was injected into a cuvette to allow for the subsequent reaction. A substantial increase in the A330 intensity was observed immediately after the contact with hematin (the first point). After 30 min of exposing the solution to ambient conditions to allow for a subsequent crosslinking reaction, the A330 intensity increased further to 0.18 (the second point). Finally, no significant crosslinking reaction was observed after 1.5 h (the third point and the points thereafter). To demonstrate the absence of hematin in the polymer solution, the injected polymer solutions were analyzed by ICP-OES to determine whether the immobilized hematin was eluted during the piston movement and/or injection of the polymer solution. The ICP-OES analysis was focused on the presence of Fe(III) due to the core iron–porphyrin coordination. The injected polymer solution exhibited iron ion concentrations below the detection limit of ICP-OES (<0.05 ppm, n = 5). These results indicate that the hematin is not eluted with the polymer solution. Thus, the resulting hydrogels are hematin-free.
Based on the aforementioned evidence (i.e., the successful immobilization of hematin and the formation of chemically active and stable catechol-derived intermediates), we attempted to prepare catalyst-free hydrogels. To prepare catechol-containing polymer hydrogels, we synthesized a catechol-tethered chitosan (CHI-C) as previously reported.14 Catechols tethered to chitosan backbones result in interesting changes in properties of the CHI-C when solubilized in neutral solutions, which provides a good platform for preparing adhesive hydrogels.10,14 Furthermore, the amine groups in the chitosan backbone provide a number of Michael addition sites, facilitating hydrogel formation. Fig. 2a shows schematic illustrations of the in situ formation of CHI-C hydrogels by interfacial chemical activation. The CHI-C (2 wt%) was dissolved in a phosphate buffered saline (PBS, pH 7.0) with 100 mM H2O2. The mixture solution was applied in a syringe, and piston movement was performed for 1 min to increase the interfacial contact between the polymer solution and the immobilized hematin. Within 1 min, the polymer solution exhibited increased viscosity so that one could inject the solution with ease (gray, Fig. 2a), which later transformed to a hydrogel (brown).
 | ||
| Fig. 2 (a) Schematic representation of the CHI-C hydrogel formation caused by an interfacial hematin-triggered reaction within the syringe. Catechol groups tethered along the chitosan backbone were activated by hematin, and then the activated catechol groups reacted with other activated moieties. (b) Photographic images of CHI-C hydrogels prepared by mixing chitosan and H2O2 in the hematin-immobilized syringe for 1 min. The CHI-C solution was immediately transformed to a viscous solution after injection (left) and gradually solidified upon exposure to ambient conditions after 3 min (middle). After 10 min of ambient exposure, the CHI-C formed three-dimensional, bulk hydrogels (right). (c) Elastic (G′, red) and viscous (G′′, black) modulus changed in the CHI-C hydrogels as a function of time after 1 min of contact with the immobilized hematin within the syringe. (d) A frequency sweep experiment, monitoring the viscoelasticity of the hydrogel by collecting the elastic modulus values. The CHI-C alone (black) and the hydrogel after 10 min (blue), 20 min (green), 30 min (purple), and 60 min (red) of ambient exposure following a 1 min contact with the hematin. | ||
For quantitative analysis, we performed rheometric studies to monitor the gelation time of the polymer solution. The elastic modulus (G′, Pa) (red circle, Fig. 2c) of the CHI-C solution was dramatically increased from 0 kPa after injection to 7.3 kPa after 1 h. The gelation point (G′ > G′′) was found to occur at approximately 20 min after injection (Fig. 2c). This finding indicates that the chemically activated catecholquinone/SFR moieties do not lose their ability to covalently crosslink with the amine and catechol groups, even in the absence of catalysts. A frequency sweep of the hydrogel as a function of time was also performed to monitor the physical properties of the injected polymer solution (Fig. 2d). The five data points of the frequency sweep experiments correspond to changes in the elastic moduli. First, the CHI-C solution showed viscoelastic behavior after injection (i.e., t = 0); the elastic moduli then increased as the rotational frequency increased (black triangle). During crosslinking, the viscoelastic property is lost, and the material becomes purely elastic (i.e., a hydrogel) (diamond to circle, Fig. 2d). Finally, after 1 h of exposure to ambient conditions, a constant elastic modulus of the CHI-C hydrogels was observed, regardless of the frequency. We also investigated the biocompatibility of the catalyst-free CHI-C hydrogels because of the toxicity caused by the remaining H2O2 with the hydrogel. The cell viability (NIH3T3) test exhibited good cell viability (>95%, Fig. S2, ESI†).
In summary, we have demonstrated, for the first time, a method for preparing catalyst-free hydrogels by utilizing surface-immobilized hematin and catechol-conjugated polymers. Through simple physical contact with immobilized hematin, the catechol-containing polymers were activated to produce chemically active intermediates such as the catecholquinone/SFR in a bulk solution. These intermediates later undergo inter-molecular reactions, forming C–C/C–O/C–N couplings in aromatic rings. Additionally, the injected solutions do not contain hematin during the injection procedure indicating that unpredictable toxicity resulting from the incorporation of the catalyst was reduced. Our results suggest that catecholic hydrogels can be formed by interfacial catalytic activation of the immobilized catalysts inside a syringe surface that can be a novel platform to prepare the catalyst-free hydrogels.
This study is supported by the National Research Foundation, the Molecular-level Interface Research Center (2009-0083525), the World Premier Material Development Program (10037915) from The Ministry of Knowledge and Economy, and the Future Therapeutic Materials Development (A120170) from The Ministry of Health and Welfare. This work is supported by the POSCO Chung-Am fellowship.
Notes and references
- (a) A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CAS; (b) J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CAS; (c) T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CAS; (d) R. J. Russell and M. V. Pishko, Anal. Chem., 1999, 71, 3126–3132 CAS; (e) D. Seliktar, Science, 2012, 336, 1124–1128 CAS.
- (a) T. Chen, H. D. Embree, E. M. Brown, M. M. Taylor and G. F. Payne, Biomaterials, 2003, 24, 2831–2841 CAS; (b) J. J. Sperinde and L. G. Griffith, Macromolecules, 1997, 30, 5255–5264 CrossRef CAS; (c) B. P. Lee, J. L. Dalsin and P. B. Messersmith, Biomacromolecules, 2002, 3, 1038–1047 CAS; (d) R. Jin, C. Hiemstra, Z. Zhong and J. Feijen, Biomaterials, 2007, 28, 2791–2800 CrossRef CAS PubMed.
- U.S. National Library of Medicine, Drug Information Portal Home Page, http://druginfo.nlm.nih.gov.
- (a) A. Uliniuc, M. Popa, T. Hamaide and M. Dobromir, Cellul. Chem. Technol., 2012, 46, 1–11 CAS; (b) D. A. Ossipov and J. Hilborn, Macromolecules, 2006, 39, 1709–1718 CrossRef CAS; (c) Y. L. Cheng, C. L. He, C. S. Xiao, J. X. Ding, H. T. Cui, X. L. Zhuang and X. S. Chen, Biomacromolecules, 2013, 14, 468–475 CAS; (d) M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774–2776 CAS.
- (a) M. d'Ischia, A. Napolitano, A. Pezzella, P. Meredith and T. Sarna, Angew. Chem., Int. Ed., 2009, 48, 3914–3921 CrossRef CAS PubMed; (b) A. Pezzella, O. Crescenzi, A. Natangelo, L. Panzella, A. Napolitano, S. Navaratnam, R. Edge, E. J. Land, V. Barone and M. d'Ischia, J. Org. Chem., 2007, 72, 1595–1603 CAS; (c) S. Parvez, M. Kang, H. S. Chung and H. Bae, Phytother. Res., 2007, 21, 805–816 CAS.
- (a) J. A. Akkara, J. Z. Wang, D. P. Yang and K. E. Gonsalves, Macromolecules, 2000, 33, 2377–2382 CAS; (b) S. Sakai, K. Moriyama, K. Taguchi and K. Kawakami, Biomacromolecules, 2010, 11, 2179–2183 CAS; (c) P. A. Riley, Ann. N. Y. Acad. Sci., 1988, 551, 111–120 CrossRef CAS; (d) J. H. Waite, N. H. Andersen, S. Jewhurst and C. J. Sun, J. Adhes., 2005, 81, 297–317 CAS; (e) M. E. Yu, J. Y. Hwang and T. J. Deming, J. Am. Chem. Soc., 1999, 121, 5825–5826 CAS; (f) B. Liu, L. Burdine and T. Kodadek, J. Am. Chem. Soc., 2006, 128, 15228–15235 CAS; (g) B. P. Lee, J. L. Dalsin and P. B. Messersmith, Biomacromolecules, 2002, 3, 1038–1047 CAS; (h) M. Kurisawa, J. E. Chung, Y. Y. Yang, S. J. Gao and H. Uyama, Chem. Commun., 2005, 4312–4314 CAS; (i) B. Kalyanaraman, C. C. Felix and R. C. Sealy, Environ. Health Perspect., 1985, 64, 185–198 CAS.
- J. H. Ryu, Y. Lee, M. J. Do, S. D. Jo, J. S. Kim, B.-S. Kim, G.-I. Im, T. G. Park and H. Lee, Acta Biomater., 2014, 10, 224–233 CAS.
- (a) A. F. G. Slater, W. J. Swiggard, B. R. Orton, W. D. Flitter, D. E. Goldberg, A. Cerami and G. B. Henderson, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 325–329 CAS; (b) D. L. Lips, C. A. Pierach and P. S. Edwards, Toxicol. Lett., 1978, 2, 329–332 CAS; (c) C. D. Fitch, R. Chevli, P. Kanja-nanggulpan, P. Dutta, K. Chevli and A. C. Chou, Blood, 1983, 62, 1165–1168 CAS.
- (a) B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CAS; (b) J. Berger, M. Reist, J. M. Mayer, O. Felt, N. A. Peppas and R. Gurny, Eur. J. Pharm. Biopharm., 2004, 57, 19–34 CAS.
- K. Kim, J. H. Ryu, D. Y. Lee and H. Lee, Biomater. Sci., 2013, 1, 783–790 CAS.
- (a) H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CAS; (b) S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CAS.
- (a) E. B. Samson, B. S. Goldschmidt, P. J. Whiteside, A. S. Sudduth, J. R. Custer, B. Beerntsen and J. A. Viator, J. Opt., 2012, 14, 065302 Search PubMed; (b) S. Nagarajan, R. Nagarajan, R. Tyagi, J. Kumar, F. F. Bruno and L. A. Samuelson, J. Macromol. Sci., Part A, 2008, 45, 951–956 Search PubMed.
- (a) J. J. Wilker, Curr. Opin. Chem. Biol., 2010, 14, 276–283 CAS; (b) D. G. Anderson, S. V. Mariappan, G. R. Buettner and J. A. Doorn, J. Biol. Chem., 2011, 286, 26978–26986 CAS.
- J. H. Ryu, Y. Lee, W. H. Kong, T. G. Kim, T. G. Park and H. Lee, Biomacromolecules, 2011, 12, 2653–2659 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed information for synthesis and experimental procedures. See DOI: 10.1039/c3cc49043b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |