 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A simple route to azaborinylphosphines: isoelectronic B–N analogues of arylphosphine ligands†
Jonathan A.
Bailey
,
Mairi F.
Haddow
and
Paul G.
Pringle
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: paul.pringle@bristol.ac.uk; Fax: +44 (0)117 929 0509; Tel: +44 (0)117 928 8114
First published on 9th December 2013
Abstract
Azaborinylphosphines are readily prepared by the reaction of silylphosphines with a chloroborane under mild conditions; they are shown to contain P–B bonds that are sufficiently robust to allow these ligands to be used in homogeneous catalysis.
Phosphorus(III) ligands have been a cornerstone of much coordination chemistry and homogeneous catalysis for half a century.1 The strength of P–C, P–N and P–O bonds makes ligands based on these bonds robust and amenable to design, in terms of stereoelectronic properties, to an unsurpassed level of finesse. As a result, a cornucopia of applications in catalysis has emerged based on P-donor complexes.
Ligands containing P–F and P–B bonds have been underdeveloped despite the fundamental interest arising from the extremes of electron density that these bonds might be predicted to confer on the P atom. Recently, fluorophosphines (e.g. fluorophobane I)2 and fluorophosphites (e.g.II),3 which contain inert P–F bonds, have not only been reported but have been shown to have potentially commercial applications in hydroformylation4 or hydrocyanation5 catalysis.
The low electronegativity of boron (B: 2.0; P: 2.2; C: 2.5) combined with the electron deficiency of BX3 compounds makes the coordination chemistry of borylphosphines (phosphinoboranes) fascinating to explore. The reactive P–B bond can be stabilised by: (i) having π-donating NR2 substituents on the B as in (III);6 (ii) having very bulky aryl substituents on the B as in (IV);7 (iii) enhancing the P–B π-bonding by having electron-accepting groups on B and electron-donating groups on P as in (V);8,9 (iv) incorporating the B into a carborane as in (VI).10 The proposition that the coordination chemistry of borylphosphines should be academically interesting has been validated by the report of the remarkably strong σ-donor capacity of (VI)10 and the Pt(0) complex of (V) which is the first example of side-on, alkene-like coordination of a phosphinoborane.9 None of the previously reported phosphinoboranes have been shown to be ligands that support homogeneous catalysis.
We are interested in the preparation of phosphorus ligands in which a BN formally replaces an isoelectronic CC group in an aromatic ring to produce azaborinyl analogues of aryl phosphines and compare their properties. It was predicted that the azaborinylphosphines would be stabilised by the aromaticity of the BN-containing ring and therefore may be potentially useful as ligands. Here we describe a simple, high-yielding route to azaborinylphosphines, compare their structural and electronic properties with the parent arylphosphines, and show that these azaborinylphosphines form stable complexes with Rh which are catalysts for hydrogenation.
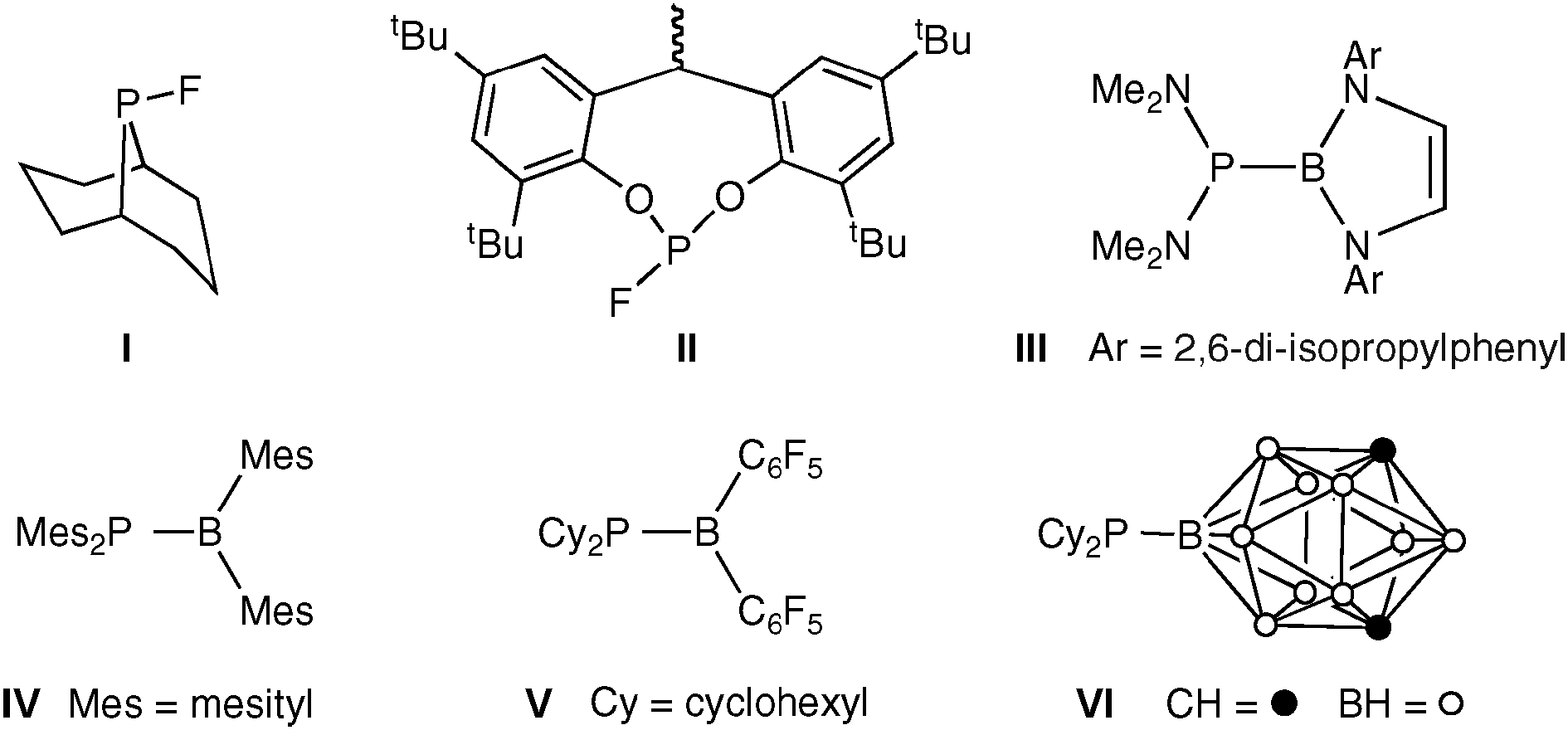
The first targets were phosphines L1a and L1b derived from the BN analogue of phenanthrene and these were to be compared with the all-carbon analogues L2a and L2b. The readily prepared6,7 chloroborane 1 was the proposed precursor to L1a and L1b. However, 1 did not react with the secondary phosphines HPiPr2 or HPPhob in the presence of amine bases (NEt3 or NEtiPr2) and treatment of 1 with LiPR2 led to the formation of secondary phosphine R2PH (∼70%, by 31P NMR spectroscopy) and a mixture of several B-containing products (according to 11B NMR spectroscopy). It was reasoned that a non-basic source of PR2 was required and so we turned our attention to silylphosphines. The chloroborane 1 reacts with Me3SiPiPr2 or Me3SiPPhob‡ rapidly and quantitatively under mild conditions to give the desired azaborinylphosphines L1a and L1b (Scheme 1). The facility of these reactions was a surprise since it had been reported11 that formation of [Et2P–BZ2]n from the reaction of Et2PSiMe3 with ClBZ2 (Z = F, Cl, Br, OnBu or nPr) required several hours at high temperatures (120–180 °C) and in the case of Z = NMe2, the route was unsuccessful.

 | ||
| Scheme 1 Synthesis of the azaborinylphosphines and their carbon analogues. | ||
The phenanthryl analogues L2a and L2b are new compounds‡ which have been prepared from the lithio reagent according to Scheme 1 which is a modification of the method reported for the preparation of the 9-diphenylphosphinophenanthrene.12 The broad 31P NMR signals for the azaborinylphosphines are at −49.1 ppm for L1a and −64.5 ppm for L1b which are both ∼40 ppm to high field of the phenanthrene analogues L2a (−7.2 ppm) and L2b (−23.8 ppm) but close in chemical shift to the silylphosphine precursors iPr2PSiMe3 (−44.0 ppm) and PhobPSiMe3 (−67.4 ppm). The similarity of the δP values for the analogous P–B and P–Si compounds is an NMR manifestation of the B–Si diagonal relationship.
In order to compare the donor properties of ligands L1a and L1b with L2a and L2b, the corresponding trans-[RhCl(CO)L2] complexes 3a,b and 4a,b were prepared and their crystal structures determined (Fig. 1 and 2). Complexes 3a and 3b are the first examples of rhodium complexes of phosphinoboranes.13 The structures of azaborinylphosphine complex 3a and its phenanthrylphosphine analogue 4a are isomorphous and all the Rh–ligand bonds are essentially the same lengths (Fig. 1). The conformations of the ligands in complexes 3b and 4b featuring the PhobP group are quite different from their iPr2P analogues 3a and 3b. The 90° torsion angles, Rh–P–B–N in 3b and Rh–P–C–C in 4b, contrast with the ca. 140° for these angles in the analogues 3a and 4a. One plausible explanation for this difference is that the phobane ligands are prevented from adopting a 140° conformation because of unfavourable clashing of the H on the C10 or N10 with the axial protons within the phosphabicycle that would result; the rigidity of the phobane makes these clashes unavoidable whereas the greater rotational freedom of the isopropyl groups of the iPr2P group allows the 140° conformer to be adopted. Another striking structural difference is that the PhobP complexes 3b and 4b exhibit extensive π-stacking between the aromatic units whereas the iPr2P analogues 3a and 4a do not; this is illustrated in the crystal packing structures for 3b and 4b shown in Fig. 3.

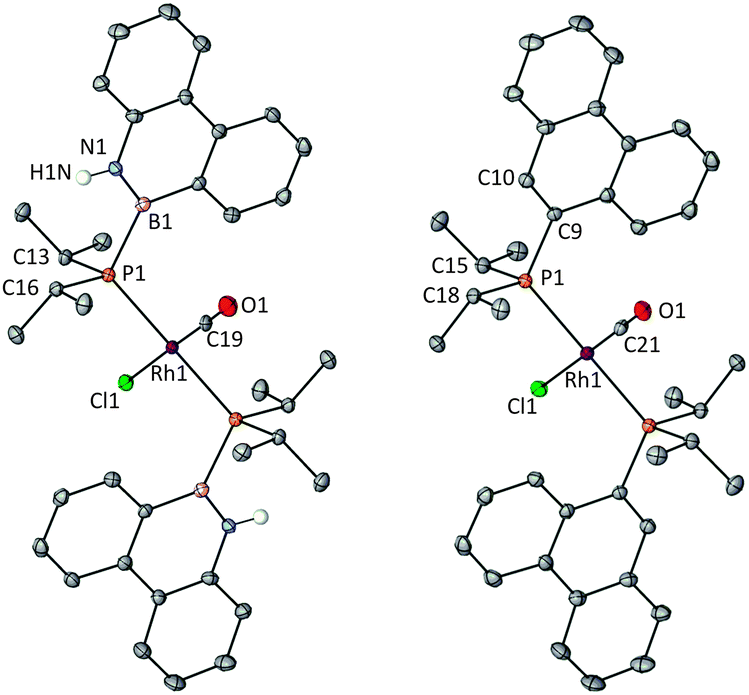 | ||
| Fig. 1 Thermal ellipsoid (50% probability) plot of 3a (left) and 4a (right), omitting all C–H hydrogen atoms. Selected bond lengths [Å] and angles [°] for 3a: Rh1–P1 2.3407(6), C19–O1 1.171(6), N1–B1 1.406(3), P1–B1 1.957(3), Rh1–P1–B1–N1 143.64. For 4a: Rh1–P1 2.3369(6), C21–O1 1.147(7), P1–C9 1.843(2), Rh1–P1–C9–C10 142.61. | ||
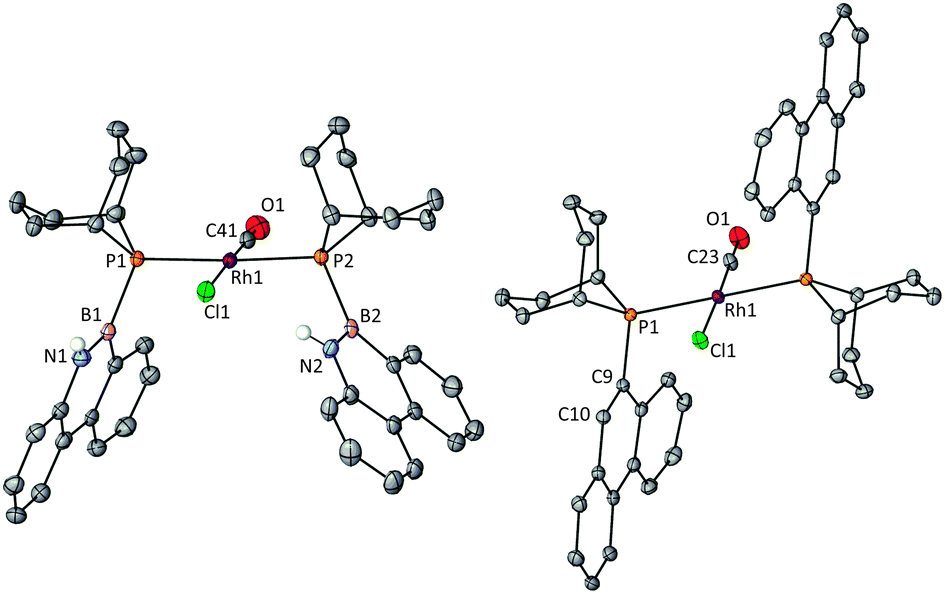 | ||
| Fig. 2 Thermal ellipsoid (50% probability) plot of 3b (left) and 4b (right), omitting all C–H hydrogens. Selected bond lengths [Å] and angles [°] for 3b: P1–Rh1 2.3296(5), P2–Rh1 2.3304(5), C41–O1 1.078(3), B1–P1 1.941(2), B2–P2 1.962(3), B1–N1 1.404(3), B2–N2 1.408(4), Rh1–P1–B1–N1 96.50, Rh1–P2–B2–N2 −71.78. For 4b: Rh1–P1 2.3313(3), O1–C23 1.155(4), P1–C9 1.8281(11), Rh1–P1–C9–C10 98.72. | ||
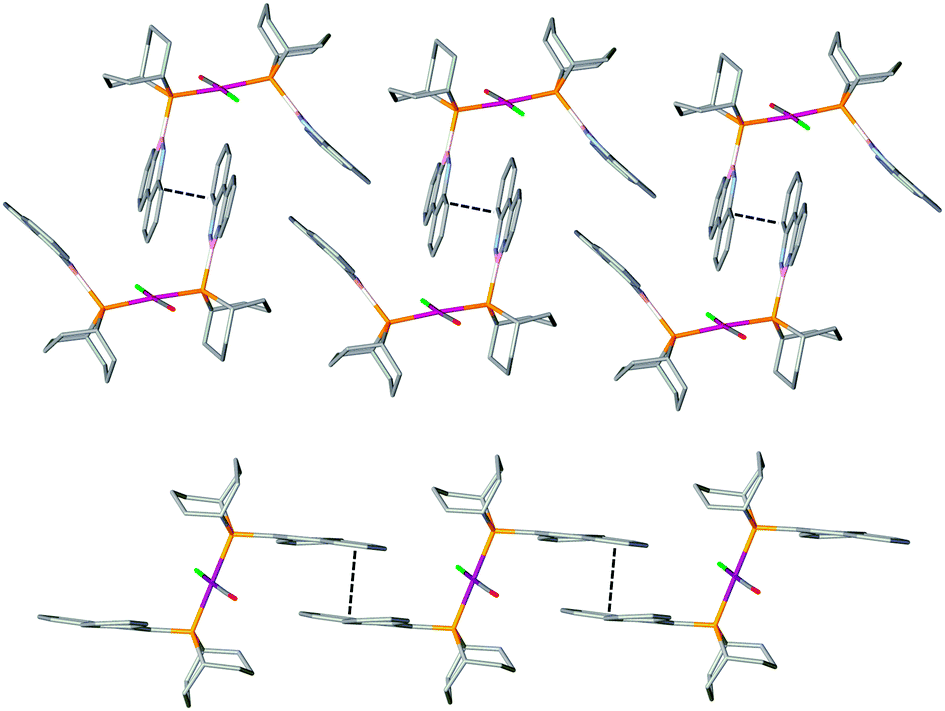 | ||
| Fig. 3 Crystal packing structures of 3b (top) and 4b (bottom). | ||
The ν(CO) values for 3a,b and 4a,b show that the BN ligands L1a and L1b are more electron-donating than their CC counterparts L2a and L2b (Table 1), as was expected from the prediction that the boryl substituent would make the P-donor electron-rich.10 It was hoped that the borylphosphines L1a and L1b would be members of a new class of ligands for catalysis. Therefore [Rh(nbd)2][BF4] was reacted with 2 equivalents of L1a and L1b to generate single products, assigned the structures 5a and 5b on the basis of their 31P NMR spectra (see Table 1 for the data) which were tested for the catalytic hydrogenation of cyclohexene. The catalytic results obtained for 5a and 5b and the phenanthryl analogues 6a and 6b are collected in Table 1. In all cases 100% conversion of the cyclohexene was observed. It is clear from Table 1 that (a) the borylphosphine complexes 5a and 5b are more efficient catalysts than their phenanthrene phosphine counterparts 6a and 6b; (b) the bicyclic phosphine complexes 5b and 6b are more efficient catalysts than their acyclic counterparts 5a and 6a.
| Complex | ν(CO)/cm−1 | δ P/ppm | J RhP/Hz | Rel. TOFe |
|---|---|---|---|---|
| a Measured in CH2Cl2. b Measured in CD2Cl2. c Cyclohexene hydrogenation carried out in CD2Cl2 with ∼10 mg cyclohexene, 5 mol% catalyst and 2 bar H2. Conversions were monitored by 1H NMR spectroscopy. d Estimated from weak NMR signals. e Relative TOF were estimated from rates of product formation at low conversion. The value of TOF for 6a was ca. 8 h−1. | ||||
| 3a | 1951 | 0.8 | ∼120d | — |
| 3b | 1946 | −17.6 | ∼120d | — |
| 4a | 1966 | 33.8 | 121 | — |
| 4b | 1951 | 15.3 | 118 | — |
| 5a | — | −20.6 | 129 | 15 |
| 5b | — | −30.7 | 130 | ≥30 |
| 6a | — | 19.3 | 146 | 1 |
| 6b | — | −2.0 | 152 | 12 |
Addition of water (ca. 0.1% by volume) to CH2Cl2 solutions of the borylphosphines L1a or L1b led to 100% hydrolysis to R2PH and 10-hydroxy-9,10-azabora-phenanthrene in less than 5 min. In similar aqueous media, the Rh-coordinated L1b in 5b was significantly more resistant to hydrolysis than the free ligand with 75% of the complex remaining after 5 min. Nevertheless it was possible that the favourable catalytic results given in Table 1 for the borylphosphine complexes were the result of hydrolysis by adventitious water giving a highly active catalyst. To refute this proposition, the hydrogenation was carried out using a sample of 5b which had been deliberately hydrolysed prior to the catalysis. Using this degraded catalyst, after 5 min, only ca. 5% cyclohexene conversion was observed, compared to the 100% conversion when pure 5b was used as the catalyst.
In conclusion, it has been shown that the reaction of silyl phosphines iPr2PSiMe3 and PhobPSiMe3 with 9-aza-10-bora-10-chlorophenanthrene afforded the corresponding azaborinyl phosphines under very mild conditions. The generality of this P–Si → P–B conversion for the synthesis of BN analogues of arylphosphines is currently under investigation. Moreover azaborinyl phosphines have proved to be competent ligands for rhodium-catalysed hydrogenation and indeed produce more active catalysts than their arylphosphine analogues.
We thank the EPSRC and COST action CM0802 “PhoSciNet” for supporting this work and Dr Thomas R. Leonard for the sample of PhobPSiMe3.
Notes and references
- P. W. N. M. Van Leeuwen, Homogeneous Catalysis, Understanding the Art, Kluwer Academic Publishers, Dordrecht, Boston, London, 2004 Search PubMed.
- N. Fey, M. Garland, J. P. Hopewell, C. L. McMullin, S. Mastroianni, A. G. Orpen and P. G. Pringle, Angew. Chem., Int. Ed., 2012, 51, 118–122 CrossRef CAS PubMed.
- T. A. Puckette, Top. Catal., 2012, 55, 421–425 CrossRef CAS.
- T. A. Puckette, US Pat., 2007, US7301054, (to Eastman) Search PubMed; T. A. Puckette and G. E. Struck, US Pat., 1998, US5840647 (to Eastman) Search PubMed; Y.-S. Liu and J. L. Rodgers, US Pat., 2009, US 2009/071121 (to Eastman) Search PubMed.
- S. Mastroianni, P. G. Pringle, M. Garland and J. Hopewell, WO Pat., 2010, WO 2010/145960 (to Rhodia) Search PubMed.
- M. Kaaz, J. Bender, D. Förster, W. Frey, M. Nieger and D. Gudat, Dalton Trans., 2014, 43, 680–689 RSC.
- D. C. Pestana and P. P. Power, J. Am. Chem. Soc., 1991, 113, 8426–8437 CrossRef CAS.
- S. J. Geier, T. M. Gilbert and D. W. Stephan, J. Am. Chem. Soc., 2008, 130, 12632–12633 CrossRef CAS PubMed; S. J. Geier, T. M. Gilbert and D. W. Stephan, Inorg. Chem., 2010, 50, 336–344 CrossRef PubMed.
- A. Amgoune, S. Ladeira, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2012, 134, 6560–6563 CrossRef CAS PubMed.
- A. M. Spokoyny, C. D. Lewis, G. Teverovskiy and S. L. Buchwald, Organometallics, 2012, 31, 8478–8481 CrossRef CAS PubMed.
- H. Nöth and W. Schraegle, Z. Naturforsch., B: Chem. Sci., 1961, 16, 473–474 Search PubMed; H. Nöth and W. Schraegle, Chem. Ber., 1965, 98, 352–362 CrossRef; R. T. Paine and H. Nöth, Chem. Rev., 1995, 95, 343–379 CrossRef.
- M. Osawa, M. Hoshino, M. Akita and T. Wada, Inorg. Chem., 2005, 44, 1157–1159 CrossRef CAS PubMed.
- A boratophosphine–rhodium complex has been reported. D. A. Hoic, W. M. Davis and G. C. Fu, J. Am. Chem. Soc., 1996, 118, 8176–8177 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Syntheses, experimental details and crystallographic data. CCDC 973285–973291. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc49000a |
| ‡ The crystal structures of PhobPSiMe3, La and L2b (as a mixture with its oxide) are described in the ESI.† |
| This journal is © The Royal Society of Chemistry 2014 |