 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural analysis and reactivity of unusual tetrahedral intermediates enabled by SmI2-mediated reduction of barbituric acids: vinylogous N-acyliminium additions to α-hydroxy-N-acyl-carbamides†
Michal
Szostak
*,
Brice
Sautier
and
David J.
Procter
*
School of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: michal.szostak@manchester.ac.uk; david.j.procter@manchester.ac.uk; Fax: +44 (0)161 2754939; Tel: +44 (0)161 2751425
First published on 12th December 2013
Abstract
Structural characterisation and reactivity of new tetrahedral intermediates based on a highly modular barbituric acid scaffold, formed via chemoselective electron transfer using the SmI2–H2O reagent, are reported. Lewis acid promoted cleavage of bicyclic α-amino alcohols affords vinylogous N-acyliminium ions, which undergo selective (>95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 1,4 over 1,2) capture with a suite of diverse nucleophiles in a practical sequence to biologically active uracil derivatives.
:5, 1,4 over 1,2) capture with a suite of diverse nucleophiles in a practical sequence to biologically active uracil derivatives.
Tetrahedral intermediates formed in the nucleophilic addition to carboxylic acid derivatives are among the most important intermediates in organic synthesis; however, only a few examples have been isolated to date due to their transient nature.1 These species have important biological implications as models for in vivo acyl-transfer reactions. Moreover, the isolation of tetrahedral intermediates is critical for structural insight into the trajectory of nucleophilic attack onto the carbonyl group and enables the development of novel synthetic equivalents for chemoselective addition reactions with carboxylic acid derivatives.2
Bicyclic uracils as direct homologues of primary nucleobases are abundant motifs in medicinal chemistry.3 Recently, derivatives of this heterocyclic scaffold have been shown to be effective as selective modulators of ionotropic glutamate receptors and exhibit activity against hepatitis C virus and HIV-1 integrase.4 While several procedures have been developed for the preparation of bicyclic uracils,5 there is a need for robust methods for the synthesis of structurally diversified analogues (Fig. 1 and 2).
 | ||
| Fig. 1 Previous and current study: synthesis, structural characterisation and reactivity of unusual tetrahedral intermediates enabled by Sm(II). | ||
 | ||
| Fig. 2 (a) Biologically-relevant uracil derivatives. (b) Examples of isolated tetrahedral intermediates (cationic and anionic adducts). | ||
Recently, we reported the preparation of unusual tetrahedral intermediates exploiting the robust nature of the barbituric acid template.6 The success of our approach relied on a polarity reversal process utilizing SmI2–H2O as a mild electron transfer reagent operating under conditions that are fully orthogonal to other single and two-electron reaction pathways.7,8 In our initial investigation, we found that barbituric acid derived hemiaminals are thermodynamically or kinetically stable, and characterized by the beginning of collapse of the tetrahedral intermediate either by elimination of the hydroxyl group to give acyliminiums or by opening of the C–N(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group to furnish alicyclic ureids. However, these initially structurally-characterised examples were limited to specific cases and did not allow for translation of structural information into reactivity that would allow the hemiaminals to be used to access valuable uracil analogues.
O) group to furnish alicyclic ureids. However, these initially structurally-characterised examples were limited to specific cases and did not allow for translation of structural information into reactivity that would allow the hemiaminals to be used to access valuable uracil analogues.
Herein, we report a study on the structural characterisation of novel tetrahedral intermediates derived from the barbituric acid scaffold that demonstrates the thermodynamic stability of the α-amino alcohol function in both six-membered and heterobicyclic scaffolds. We apply these structural findings to develop a highly divergent process for the synthesis of 6-substituted-uracils via nucleophilic capture of N-acyliminium ions9 that proceeds with exclusive (>95:5, 1,4 over 1,2) selectivity and demonstrate that the solid state characteristics of these hemiaminals are reflected by their solution properties. Overall, these studies set the stage for the synthesis of a wide range of diverse nitrogen-containing heterocycles and tetrahedral intermediates from cyclic scaffolds (Fig. 3).10
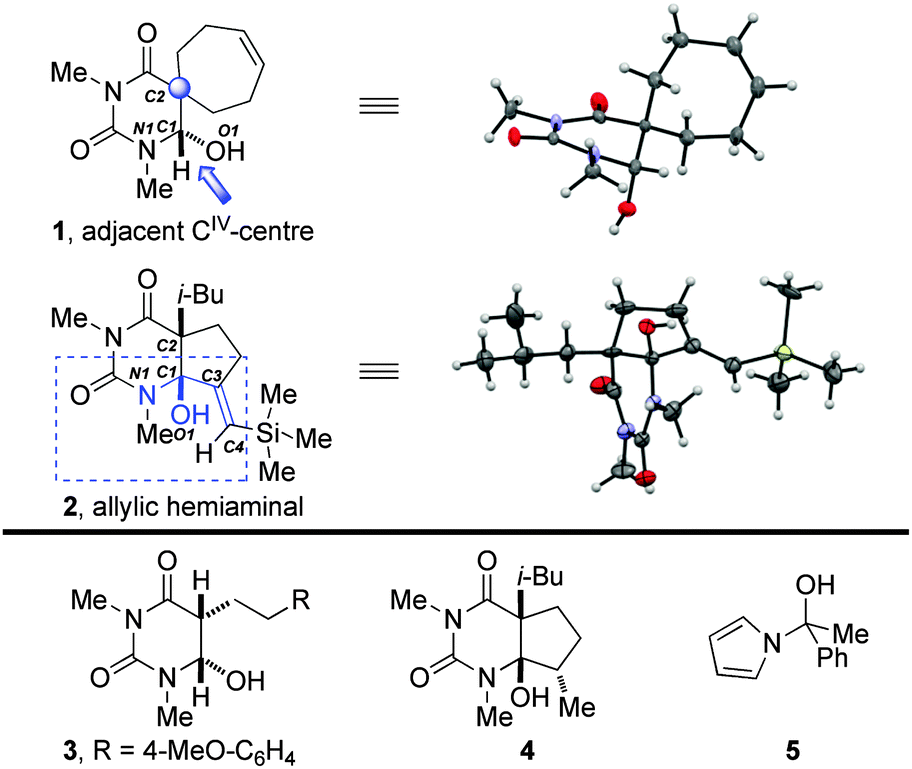 | ||
| Fig. 3 X-ray structures of 1 and 2. Structures of related compounds 3–5. | ||
The barbiturate template serves as a robust platform for the stabilization of otherwise short-lived, high-energy tetrahedral intermediates due to a combination of the stabilizing Nlp → π*CO conjugation and the scaffolding effect of the six-membered ring.6 Whereas steric effects were previously used to push the Nlp and Olp out of conjugation with the corresponding σ*C–O and σ*C–N bonds, we considered that a geminal substitution of the adjacent C2–carbon atom would affect the delocalization of Nlp and Olp in the α-amino alcohol moiety and provide insight into the extent of stabilization by the scaffold. After extensive investigation, we found that a very sterically-hindered spiro-cyclic hemiaminal 1, which was readily obtained using our recently reported reduction of barbituric acids by SmI2–H2O,6 can be examined by X-ray crystallography (recryst. from acetone/water, 50:1 v/v, mp = 136–138 °C).
Structural parameters of 1 relevant to the geometry at the anomeric centre along with bond lengths and angles of related hemiaminals are presented in Table 1. The X-ray crystal structure of 1 reveals that the C1–O1 bond (1.407 Å) is shorter than the average Csp3–O bond (1.432 Å), while the length of the N1–C1 bond is 1.463 Å, which corresponds to a typical Csp3–N bond (1.469 Å). The C1–C2 bond length of 1.522 Å is slightly shorter than the average Csp3–Csp3 bond (1.530 Å). The torsion angle between Nlp and C1–O1 of 192.2° indicates a reasonably good Nlp → σ*C–O interaction. In addition, there is a good overlap between the O1lp1 and the N1–C1 bond (∼164°) and between the O1lp2 and the C1–H bond (∼197°). These structural features indicate that the stability of the α-amino alcohol function in geminally substituted hemiaminals is thermodynamic in origin (i.e. it involves equilibrium between cyclic and alicyclic ureides as well as between hemiaminal and the corresponding iminium), thus suggesting that these systems could be applied towards generation of synthetically useful iminium ions.9
| Entry | Aminal | C1–O1 | N1–C1 | C1–C2 | Nlp → σ*C1–O1 | Olp1 → σ*C1–N1 | Olp2 → σ*C1–H/C3 |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 1.407 | 1.463 | 1.522 | 12.2 | 163.5 | 197.4 |
| 2 | 2 | 1.422 | 1.470 | 1.530 | 62.7 | 165.1 | 171.0 |
| 3 | 3 | 1.413 | 1.461 | 1.518 | 7.4 | 131.5 | 131.7 |
| 4 | 4 | 1.407 | 1.466 | 1.549 | 57.4 | 171.0 | 55.1 |
| 5 | 5 | 1.412 | 1.478 | 1.528 | 46.7 | 182.0 | 179.1 |
To gain insight into the extent of electronic stabilization on the Nlp and Olp donation, we considered a barbituric acid hemiaminal 2 featuring an unusual allylic α-amino alcohol moiety. The X-ray structure of 2 (recryst. from acetone/water, 50:1 v/v, mp = 156–158 °C) is consistent with the thermodynamic stability. The torsion angle between Nlp and C1–O1 of 62.7° is in agreement with the absence of Nlp → σ*C–O interactions. However, there is a good overlap between the O1lp1 and the N1–C1 bond (∼165°) and between the O1lp2 and the C1–C3 bonds (∼171°). The N1–C1–C3–C4 torsion angle of ∼20° and O1–C1–C3–C4 of ∼100° reveal co-planarity of the exo-cyclic double bond with the hemiaminal nitrogen. These structural features indicate electronic stabilization at the anomeric carbon and suggest the propensity of these hemiaminals to form extended N-acyliminium ions via hydroxyl elimination. The structural differences between 1 and 2 are further indicated by the bond lengths of the alpha hydroxyl urea moiety (1, N–C(O) bond length of 1.413 Å, CO of 1.224 Å; 2, N–C(O) = 1.386 Å, CO = 1.231 Å), consistent with an enhanced Nlp → σ*C–O donation in 1.
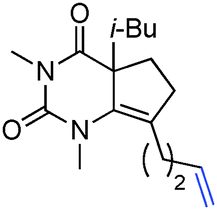
Having identified structural features contributing to the stability of these tetrahedral intermediates, we next investigated their synthetic utility. In particular, we recognized that intermediates analogous to 2 could function as vinylogous N-acyliminium equivalents whose electrophilic character is revealed under suitably acidic conditions. We were delighted to find that upon exposure of 6a to mildly Lewis acidic conditions in the presence of allyltrimethylsilane highly regioselective formation of N-Ac-urea-enamide 7a occurred in excellent yield (Table 2, entry 1). Under these reaction conditions the 1,2-addition product was not observed.11 A broad range of nucleophiles is suitable for this 1,4-addition. The addition of small nucleophiles such as nitrile and azide occurred with excellent regiocontrol (entries 3 and 4). In the latter case loss of N2 occurred, furnishing an α,β-unsaturated aldehyde bearing a vinylogous enamide. Propargyl and allenylsilane nucleophiles gave products with π systems ready for elaboration (entries 5 and 6). An electron deficient allylsilane afforded the product containing a halide functional handle (entry 7). Most remarkably, the addition of a very sterically-hindered ketene acetal Me2CC(OMe)(OSiMe3) delivered a methyl ester bearing an adjacent quaternary centre with full control of regioselectivity (entry 8). Overall, these studies demonstrate that barbituric acid derived tetrahedral intermediates afford diverse bicyclic uracils that would be very difficult to prepare using other currently available methods.
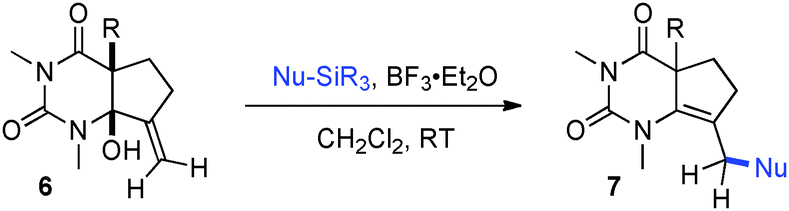










Spectroscopic studies lend additional support to the reactive properties of the α-amino alcohol moiety (Table 3 and ESI†). The solution of 1 in acetone shows a 13C NMR peak at 84.6 ppm, which moves downfield by ca. 10 ppm in bicyclic hemiaminals such as 2, consistent with deshielding of the anomeric carbon and decomposition via C–N cleavage as the predominant pathway. IR ν(CO) frequencies indicate that the N-Ac carbonyl absorbs in the region corresponding to saturated uracil derivatives (ca. 1710 cm−1), while the carbonyl group attached to the α-amino alcohol exhibits the reactive properties of an urea (bicyclic aminals, ca. 1645 cm−1) or an imide (monocyclic six-membered aminals, ca. 1658 cm−1). Thus, the spectroscopic properties correlate well with the structural stability, which decreases in the order six-membered > bicyclic hemiaminals.
| Entry | Aminal | δ 13C–OH |
δ
13CO1 |
δ
13CO2 |
Δδ13C–OH |
νCO1 |
νCO2 |
|---|---|---|---|---|---|---|---|
|
a
δ
13C–OH and δ13CO [ppm], measured at 100 MHz in CD3C(O)CD3. Δδ13C–OH = [δ13C–OH − 80.6]. νCO [cm−1].
|
|||||||
| 1 | 1 | 84.6 | 153.7 | 175.0 | 4.0 | 1658 | 1709 |
| 2 | 2 | 93.2 | 153.6 | 174.2 | 12.6 | 1645 | 1709 |
| 3 | 3 | 80.6 | 154.1 | 171.5 | — | 1657 | 1711 |
| 4 | 4 | 95.4 | 153.1 | 173.9 | 14.8 | 1644 | 1704 |
In summary, tetrahedral intermediates derived from barbituric acids via electron transfer using SmI2–H2O indicate the tendency to eliminate hydroxyl and/or N–(CO) groups to furnish N-acyliminium ions or alicyclic ureids, which is consistent with the thermodynamic stability of α-amino alcohols embedded in six-membered uracil frameworks. Studies on the synthesis of a wide range of N-containing heterocycles and tetrahedral intermediates are ongoing and will be reported shortly.12
We thank the EPRSC for financial support and James Raftery for X-ray crystallography.
Notes and references
- For reviews, see: (a) M. Adler, S. Adler and G. Boche, J. Phys. Org. Chem., 2005, 18, 193 CrossRef CAS; (b) P. Deslongchamps, Stereoelectronic Effects in Organic Chemistry, Pergamon Press, Elmsford, NY, 1983 Search PubMed; Examples of isolated tetrahedral intermediates: (c) A. J. Kirby, I. V. Komarov and N. Feeder, J. Am. Chem. Soc., 1998, 120, 7101 CrossRef CAS; (d) C. Cox, H. Wack and T. Lectka, J. Am. Chem. Soc., 1999, 121, 7963 CrossRef CAS; (e) D. A. Evans, G. Borg and K. A. Scheidt, Angew. Chem., Int. Ed., 2002, 41, 3188 CrossRef CAS; (f) M. Szostak and J. Aubé, J. Am. Chem. Soc., 2010, 132, 2530 CrossRef CAS PubMed; (g) M. Adler, M. Marsch, N. S. Nudelman and G. Boche, Angew. Chem., Int. Ed., 1999, 38, 1261 CrossRef CAS.
- D. Seebach, Angew. Chem., Int. Ed., 2011, 50, 96 CrossRef CAS PubMed.
- (a) L. Brunton, B. Chabner and B. Knollman, Goodman and Gilman's The Pharmacological Basis of Theraupeutics, McGraw-Hill, 2010 Search PubMed; (b) V. A. Bloomfield, D. M. Crothers and I. Tinoco, Nucleic Acids: Structures, Properties and Functions, University Science Books, 2000 Search PubMed.
- Selected examples: (a) G. Sun, C. J. Fecko, R. B. Nicewonger, W. W. Webb and T. P. Begley, Org. Lett., 2006, 8, 681 CrossRef CAS PubMed; (b) S. Butini, D. S. Pickering, E. Morelli, S. S. Coccone, F. Trotta, M. De Angelis, E. Guarino, I. Fiorini, G. Campiani, E. Novellino, A. Schousboe, J. K. Christensen and S. Gemma, J. Med. Chem., 2008, 51, 6614 CrossRef CAS PubMed; (c) J. Bergman and P. V. Svensson, Tetrahedron, 2010, 66, 4601 CrossRef CAS PubMed; (d) Y. Liu, B. H. Lim, W. W. Jiang, C. A. Flentge, D. K. Hutchinson, D. L. Madigan, J. T. Randolph, R. Wagner, C. J. Maring, W. M. Kati and A. Molla, Bioorg. Med. Chem. Lett., 2012, 22, 3747 CrossRef CAS PubMed.
- J. I. Bardagi and R. A. Rossi, Org. Prep. Proced. Int., 2009, 41, 479 CrossRef CAS.
- M. Szostak, B. Sautier, M. Spain, M. Behlendorf and D. J. Procter, Angew. Chem., Int. Ed., 2013, 52, 12559 CrossRef CAS PubMed.
- Recent reviews on SmI2: (a) H. B. Kagan, Tetrahedron, 2003, 59, 10351 CrossRef CAS PubMed; (b) D. J. Edmonds, D. Johnston and D. J. Procter, Chem. Rev., 2004, 104, 3371 CrossRef CAS PubMed; (c) M. Szostak and D. J. Procter, Angew. Chem., Int. Ed., 2011, 50, 7737 CrossRef CAS PubMed; (d) M. Szostak and D. J. Procter, Angew. Chem., Int. Ed., 2012, 51, 9238 CrossRef CAS PubMed; (e) M. Szostak, M. Spain and D. J. Procter, Chem. Soc. Rev., 2013, 42, 9155 RSC.
- Recent applications of SmI2–H2O: (a) L. A. Duffy, H. Matsubara and D. J. Procter, J. Am. Chem. Soc., 2008, 130, 1136 CrossRef CAS PubMed; (b) G. Guazzelli, S. De Grazia, K. D. Collins, H. Matsubara, M. Spain and D. J. Procter, J. Am. Chem. Soc., 2009, 131, 7214 CrossRef CAS PubMed; (c) M. Szostak, M. Spain and D. J. Procter, Chem. Commun., 2011, 47, 10254 RSC; (d) M. Szostak, M. Spain, K. A. Choquette, R. A. Flowers, II and D. J. Procter, J. Am. Chem. Soc., 2013, 135, 15702 CrossRef CAS PubMed.
- (a) B. E. Maryanoff, H. C. Zhang, J. H. Cohen, I. J. Turchi and C. A. Maryanoff, Chem. Rev., 2004, 104, 1431 CrossRef CAS PubMed; (b) A. Yazici and S. G. Pyne, Synthesis, 2009, 339 CAS; (c) A. Yazici and S. G. Pyne, Synthesis, 2009, 513 CAS.
- 6-Alkoxyuracils are typically prepared by formal hydration or photocycloaddition of 5,6-DH-uracils. WebCSD search revealed no examples of simple 6-hydroxy-derivatives. (a) D. J. Abraham, T. G. Cochran and R. D. Rosenstein, J. Am. Chem. Soc., 1971, 93, 6279 CrossRef CAS; (b) F. F. Kong, B. C. Zhai and Q. H. Song, Photochem. Photobiol. Sci., 2008, 7, 1332 RSC.
- Selected examples of 1,4-additions: (a) K. Kikushima, J. C. Holder, M. Gatti and B. M. Stoltz, J. Am. Chem. Soc., 2011, 133, 6902 CrossRef CAS PubMed; (b) J. A. Jordan-Hore, J. N. Sanderson and A. L. Lee, Org. Lett., 2012, 14, 2508 CrossRef CAS PubMed; (c) M. Sidera, P. M. C. Roth, R. M. Maksymowicz and S. P. Fletcher, Angew. Chem., Int. Ed., 2013, 52, 7995 CrossRef CAS PubMed.
- For 1,2-N-acyliminium addition to geminally-substituted monocyclic analogues derived from monoreduction of barbituric acids using SmI2–H2O, see: M. Szostak, B. Sautier and D. J. Procter, Org. Lett., 2014, 16, 452 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 972704 and 972705. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc48932a |
| This journal is © The Royal Society of Chemistry 2014 |