 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microplasma-chemical synthesis and tunable real-time plasmonic responses of alloyed AuxAg1−x nanoparticles†
Tingting
Yan
a,
Xiaoxia
Zhong
*a,
Amanda Evelyn
Rider
bc,
Yi
Lu
a,
Scott A.
Furman
b and
Kostya (Ken)
Ostrikov
*bcd
aKey Laboratory for Laser Plasmas (Ministry of Education) and Department of Physics and Astronomy, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: xxzhong@sjtu.edu.cn
bCSIRO Materials Science & Engineering, Lindfield, NSW 2070, Australia
cFaculty of Science, The University of Sydney, NSW 2006, Australia
dUniversity of Technology Sydney, Broadway NSW 2007, Australia
First published on 28th January 2014
Abstract
Tunable synthesis of bimetallic AuxAg1−x alloyed nanoparticles and in situ monitoring of their plasmonic responses is presented. This is a new conceptual approach based on green and energy efficient, reactive, and highly-non-equilibrium microplasma chemistry.
Due to the tunability of their properties, metal alloyed nanoparticles (NPs) are of interest for novel applications in plasmonics, catalysis, nanomedicine, medical imaging, cancer treatment, etc.1–7 By changing the nanoparticle size, composition and/or the distribution of the elements within the NP (i.e., the internal structure – either a homogeneous composition throughout the NP, a smooth compositional gradient or a core and a number of discrete shells),8–10 different properties may be obtained. Two examples of promising bimetallic alloys include AlxFey which is a low-cost candidate for the replacement of Pd as a catalyst for heterogeneous hydrogenation11 and NixFe1−x NPs synthesized via microplasma12 for the chirality control of carbon nanotubes.
Noble metal alloyed NPs13 such as AuxAg1−x,10,14–19 are extensively investigated for plasmonics-related applications,10,17–19 catalysis20–22 and biosensing,23 as they exhibit tunable properties between the values for pure Au and pure Ag.14 The NP structure (i.e., whether it is a core–shell or a well-mixed bimetallic alloy) determines its applications. In some cases, Au core–Ag shell NPs can be transformed into AuAg alloy NPs by annealing them at 250 °C.14 However, it is still quite difficult to control the growth of AuxAg1−x NPs to ensure that it is a well-mixed, crystalline alloy from the earliest possible growth stages.
Current colloidal chemistry synthesis methods of AuxAg1−x alloy NPs involve either standard chemical approaches, i.e., reducing gold and silver salts in solution in the presence of a reducing agent such as NaBH4 and a capping agent (e.g., citrate),15 or replacement reactions, which are complex, multistep processes involving elevated temperatures (up to 100 °C) and the presence of NaBH4.16 These methods have a number of drawbacks, e.g. the toxic nature of the reducing agent and the necessity to remove capping agents which can contaminate the surface (often irreversibly) with many impurities. Moreover, complicated multi-step processes are less viable for industrial applications, and high temperatures increase the energy cost of the processes. These factors make in situ UV-Vis absorption monitoring (indispensable for controlling the NP characteristics from the earliest growth stages) exceedingly difficult, and in cases where annealing is involved, simply not possible.
We present a viable alternative for the green, energy-efficient synthesis of bimetallic alloyed nanoparticles. This is a new conceptual approach based on reactive, highly non-equilibrium plasma chemistry.24–26 Our microplasma-chemical synthesis, in particular, presents an attractive alternative to the above-mentioned methods. Microplasmas feature at least one dimension in the sub-mm range27 (see the ESI,† Section S7 for more information on the microplasma characteristics). They have been used in a range of applications from bacterial inactivation to nanoparticle synthesis both in the gas12 and liquid phases.28 In the synthesis of metal nanoparticles in the liquid phase, a microplasma is used as the cathode, a metal salt/stabilizer mixture as the electrolyte and a metal foil as the anode (see Fig. 1(a and b); ESI,† Section S1). The metal foil can either take part in the synthesis (i.e., it is oxidized, providing metal ions which are then reduced at the cathode) or be an inert electrode (i.e., Pt), with the metal salt in the electrolyte providing the material for the NP synthesis. Here we use the latter, where only the metal salt in the electrolyte contributes to the nanoparticle growth.
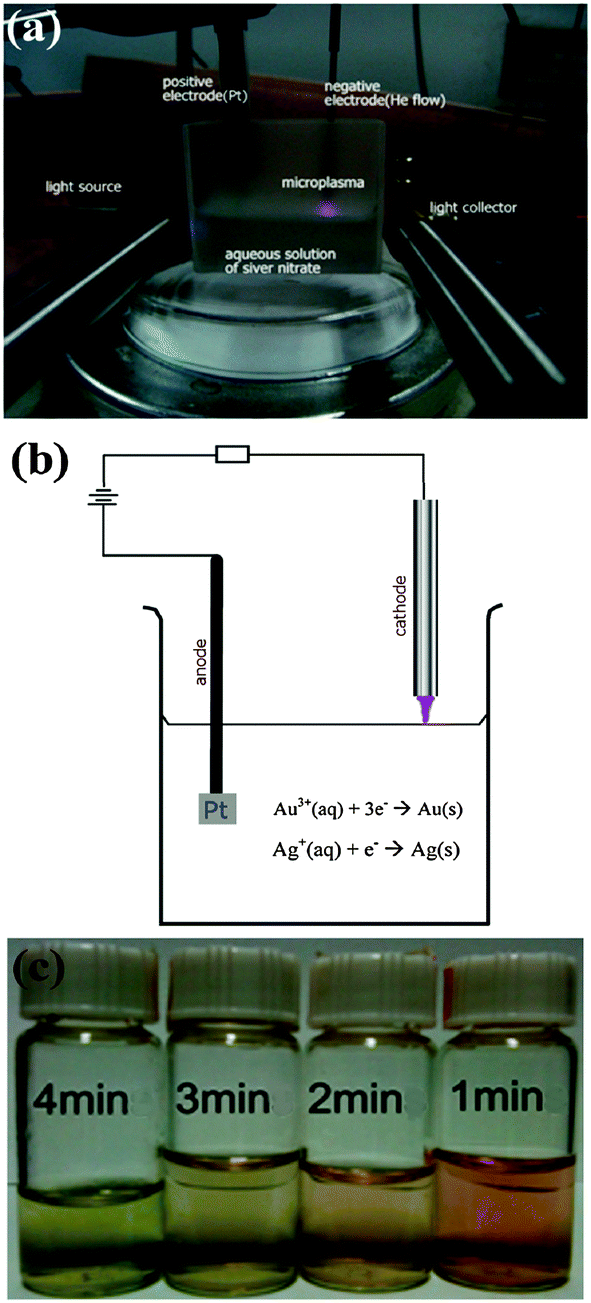 | ||
| Fig. 1 (a) Photograph of in situ monitoring set-up for the synthesis of nanoparticles, (b) the diagram of the plasma-chemical set-up used to synthesize AuxAg1−x alloy NPs. (c) The photograph of colloidal AuxAg1−x NP solutions with a processing time of 1–4 min. | ||
The advantage of using a microplasma to co-reduce Au and Ag salts (HAuCl4 and AgNO3, in this case) is that it is a comparatively green process, i.e., there is no need for reducing agents such as NaBH4, citrate or hydrazine as the plasma generates electrons (and also ions and excited radicals). Whilst the microplasma considered here is fairly chemically simple, it induces multiple reaction pathways for the synthesis of nanoparticles, from the standard dissociation and subsequent reduction reactions of metal salts that occur in colloidal synthesis as well as more exotic radical-induced reactions29–31 (see ESI,† Section S7). Microplasma-assisted electrochemical synthesis is a fast process, taking only minutes due to the energetic plasma electrons (see Fig. 1(c)), whereas many traditional chemical processes can often take hours/days. Moreover, it is a more energy efficient process than synthesis methods requiring heating (e.g., by at least a factor of 10, most likely considerably more than the common citrate method, see ESI,† Section S2). There is also a higher degree of control over the nanoparticle surface chemistry, with no undesired surface products that need to be removed, as in the case where more complex chemical mixtures are used. The main difference between the plasma-assisted method and other similar approaches (UV-, microwave-, and ultrasound-assisted) is the type of reaction-inducing agents, namely electrons, ions, and excited radicals in our case, whereas light, microwaves, and ultrasonic waves are used for UV-, microwave-, and ultrasound-assisted techniques, respectively (see ESI,† Section S8).
Here we present the liquid-phase synthesis of AuxAg1−x alloyed NPs via microplasma-induced co-reduction of metallic ions and more exotic radical induced reactions. These additional, enhanced (compared to conventional colloidal chemistry) reaction pathways, coupled with the higher energy of the plasma-generated electrons, lead to much faster formation of the AuxAg1−x nanoparticles. Moreover, this is a single-step process, under ambient conditions (i.e., room temperature and atmospheric pressure), with in situ monitoring of the NP growth easily incorporated into the experimental set-up (see Fig. 1(b)), which provides real-time information about the characteristics of the NPs as they are forming. It is shown that the composition of the NPs is tunable by varying the reaction time. Hence, the effective, non-equilibrium reactive plasma chemistry represents a viable, low-cost, and highly promising green approach for the energy-efficient and potentially scalable synthesis of a range of alloyed nanoparticles.
A transmission electron micrograph (TEM) of the synthesized Au1−xAgx NPs with a processing time of 2 minutes with the corresponding particle size distributions (PSDs, taken over a number of TEMs, fit to a Gaussian distribution) is presented in Fig. 2(a and b) [more TEMs and PSD are provided in the ESI,† Section S3]. Fig. 2(c) summarizes the data obtained from the TEMs for the 1–4 min samples. It is clear from Fig. 2(a) that the NPs are fairly uniform, both in size and shape. As seen from Fig. 2(c), generally, as processing time increases, the peak of the PSD increases from 10.1 to 13.3 nm, whilst the full width at half maximum (FWHM, roughly, a measure of the size uniformity of the NP yield) increases from 4.2 to 5.1 nm.
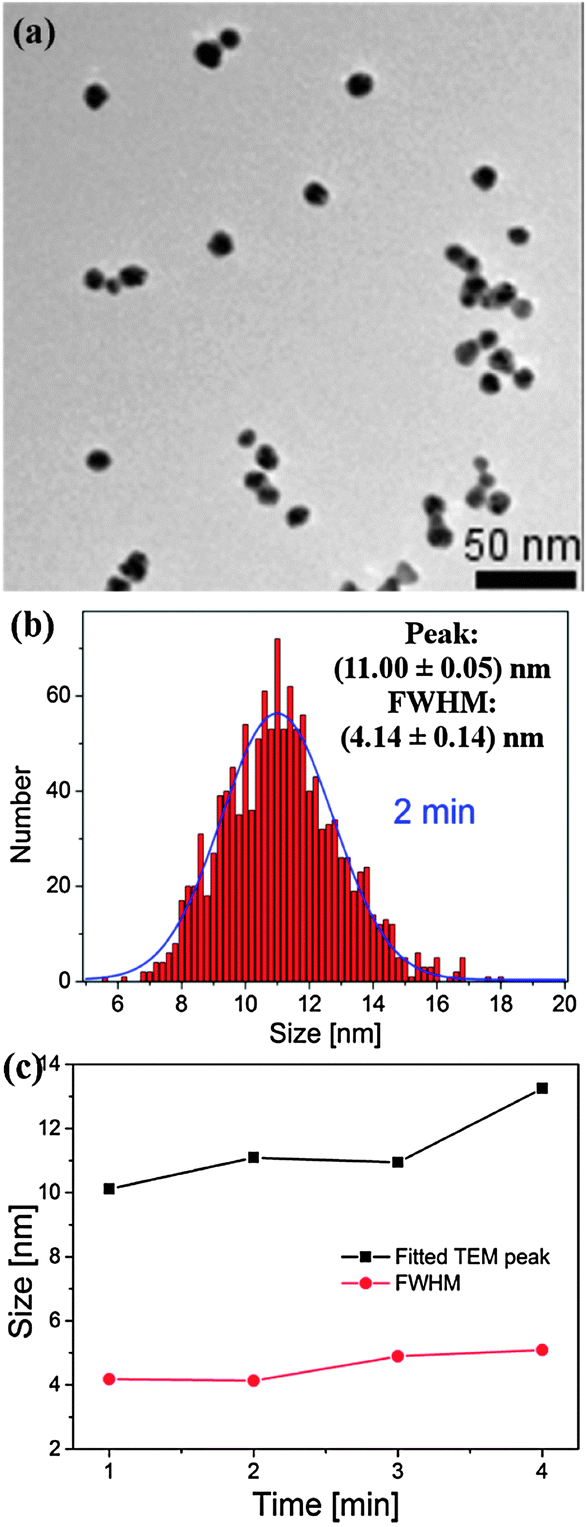 | ||
| Fig. 2 TEM (a) and PSD (b) of AuxAg1−x alloy NPs produced using a microplasma exposure of 2 min. (c) Summary of size/PSD information from TEMs. | ||
Fig. 3(a–d) show high-resolution TEMs and selected area electron diffraction (SAED) patterns, recorded on samples prepared at 1, 2, 3, and 4 min, respectively. It should be noted that no core–shell structure is discernible from the HR-TEMs (see possible interpretations in the ESI,† Section S3). These data show that it is possible to control the crystal structure by varying the growth time. For example, from the micrograph of the 4 min sample (Fig. 3(d)), it is clear that the particles are polycrystalline. The measured lattice spacing of 0.2357 nm and 0.2040 nm fall in between the Au and Ag values for (111) and (200) planes, respectively [(111): Ag = 0.2359 nm, Au = 0.2350; (200): Ag = 0.2044 nm, Au = 0.2030 nm]. We emphasize that these alloys were crystalline prior to exposure to an electron beam during HR-TEM analysis. This conclusion is also supported by X-ray diffraction spectra provided in the ESI,† Section S4.
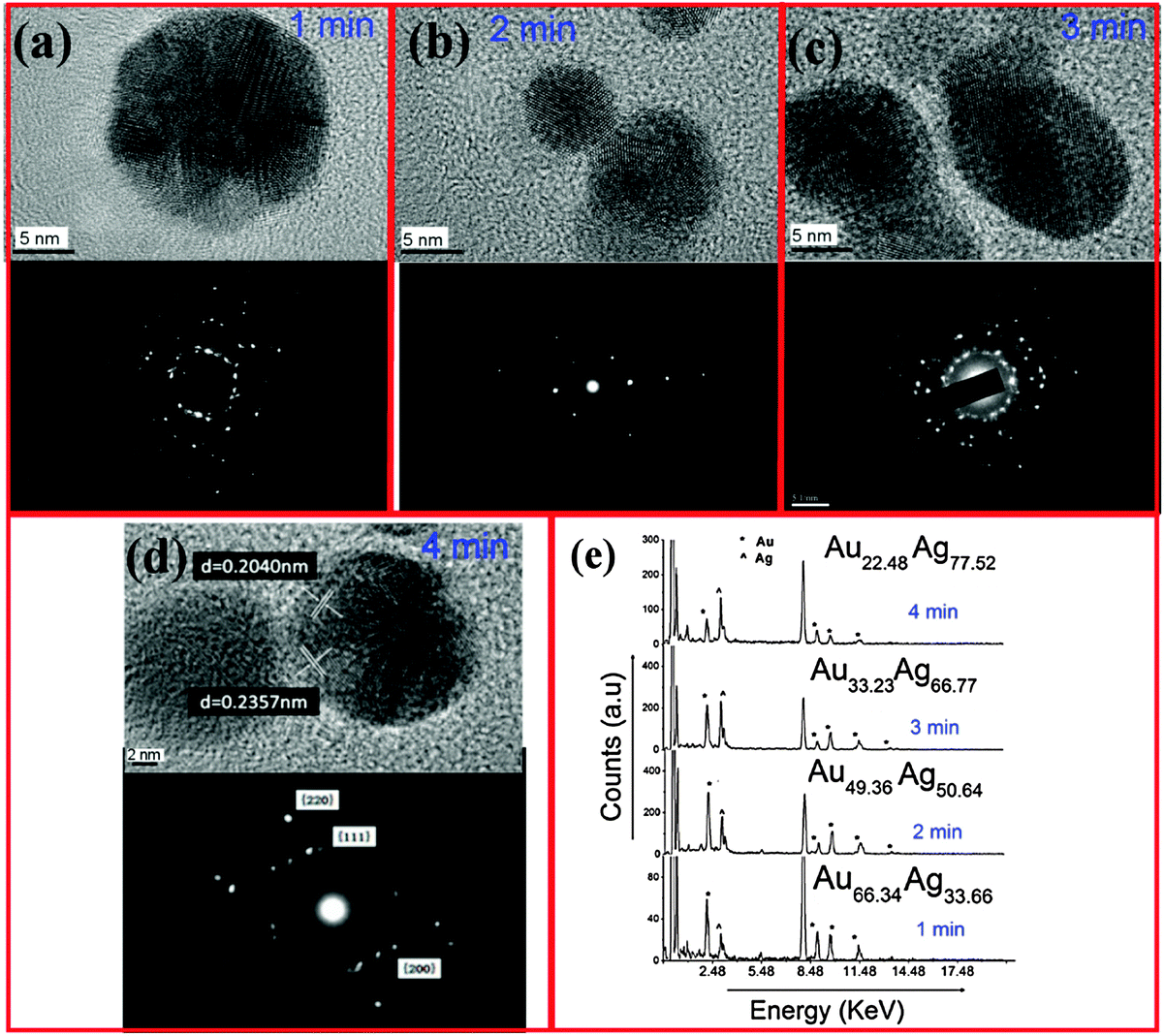 | ||
| Fig. 3 High-resolution TEMs and selected area electron diffraction patterns of AuxAg1−x alloy NPs produced using a microplasma exposure time of (a) 1 min, (b) 2 min, (c) 3 min and (d) 4 min. (e) EDX spectra of AuxAg1−x alloy NPs at different exposure times. | ||
Energy dispersive X-ray (EDX) spectra are provided in Fig. 3(e), showing that the silver content increases for longer processing times. Specifically, by comparing the ratio of the AgKα and AuKα lines in the EDX spectra, the ratio of Ag![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au was determined to be 34:66, 51:49, 67:33 and 78:22 after 1, 2, 3, and 4 min processing time, respectively. Once the NP composition is known, one can use Vegard's Law to determine the lattice constant of the alloy
:Au was determined to be 34:66, 51:49, 67:33 and 78:22 after 1, 2, 3, and 4 min processing time, respectively. Once the NP composition is known, one can use Vegard's Law to determine the lattice constant of the alloy
| aAuAg = xaAu + (1 − x)aAg, | (1) |
Let us now consider Fig. 1(c) which shows the color change of the colloidal solutions with processing time. Once the atmospheric-pressure microplasma impinges on the surface of the solution, the colorless solution first turns reddish, indicating that the AuAg nanoparticles are forming. With the increased processing time, the solution turns greenish, which indicates an increased amount of silver in the NPs. This conclusion is supported by the corresponding UV-Vis spectra (Fig. 4(a)) which show a blue shift with increased time, e.g. from 486 to 403 nm as time increases from 1 to 4 minutes. This can be explained by considering the half cell reactions for Au and Ag:
| Au3+(aq) + 3e− → Au(s) | (2) |
| Ag+(aq) + e− → Ag(s) | (3) |
The reduction potential of Au (Eo = 1.52 V) is higher than that of Ag (Eo = 0.799 V). This means that Au has a higher affinity for electrons, hence is likely to be reduced faster than the Ag ions in the solution. This is supported by the observed initial appearance of a red color in the solution. With longer processing time, the Ag ions are still reduced, even after the Au ions have been consumed (the Au being in the solution at a reduced concentration, i.e. the ratio of Au:Ag in solution was 1:4). The shape of the UV-Vis absorption spectra indicates that the produced NPs are indeed bimetallic nano-alloys.
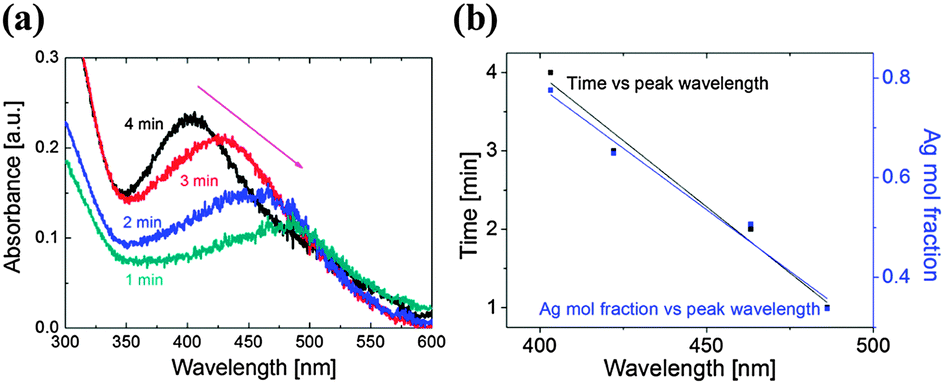 | ||
| Fig. 4 (a) UV-Vis absorbance spectra of samples produced with a 1–4 min exposure time; (b) graphs of time and the Ag mol fraction (1 − x) versus the peak UV-Vis absorbance wavelength. | ||
This interpretation is supported by the experiments of Mallin et al.,15 who noted that the appearance of 2 peaks in the UV-Vis absorption spectra is indicative of either mixed, single metal nanoparticles or, when the peaks vary in relation to each other, of core–shell nanoparticles. In contrast, a single peak can represent either single metal NPs or fairly well-mixed bimetallic nano-alloys. By considering the shape and location of the peaks shown in Fig. 4(a), we can rule out single-element NPs – thus reinforcing our conclusion that AuxAg1−x nano-alloys have been obtained.
By combining the UV-Vis data with the EDX data from Fig. 3(e) and the PSD from Fig. 2, we are able to link the UV-Vis data to processing time and composition data and hence determine that increased amounts of silver were present for a larger processing time to produce larger NPs. Fig. 4(b) shows that there is a clear linear relationship between the wavelength and the processing time, as well as the wavelength and the nanoparticle composition (see ESI,† Section S5).
Therefore, we have demonstrated that well-mixed bimetallic alloys have reliably been obtained with the x values between 22 and 66. This method is also promising for other bimetallic alloy nanoparticles (see the ESI,† Section S6 for preliminary results for AuPt NP alloys). The additional tuning of plasmonic responses in bimetallic nanoparticles (as compared to single-metal NPs) offers extra possibilities to utilize the electron charging and plasmonic effects in next-generation energy conversion devices such as dye-sensitized solar cells. Our process may be scalable by utilizing a line or a 2D array of plasma jets or dielectric barrier discharges (DBD), and eventually flow-through techniques which are currently under investigation.
The reaction rates of reduction and NP formation in the vicinity of the plasma are increased due to the influx of energetic electrons (with the equivalent ‘temperature’ in the range of tens of thousands of degrees, which is much higher compared to the electrons generated using chemical reducing agents). The energy of the electrons may be tailored by varying the plasma parameters. This may enhance the Au and Ag reduction reactions and also increase diffusion rates near the plasma.28 Plasma-generated electrons may also interact with the other species in the reaction volume (e.g., water), setting off a chain of reactions involving the creation of radicals31 (e.g., hydrogen and hydroxyl) and culminating in the reduction of metal ions, which then form nanoparticle seed nuclei (see the ESI,† Section S7 for a discussion of plasma-specific effects32,33). Hence there are multiple additional, enhanced (compared to conventional physical chemistry) reaction pathways leading to the nanoparticle synthesis. A similar process conducted using the same reagent mixture and at the same temperature but without any plasma, does not produce these nanoparticles.
In summary, we have presented a viable approach to alloyed bimetallic nanoparticle synthesis based on the effective, non-equilibrium reactive microplasma chemistry. In particular, we demonstrated a single-step, microplasma-chemical green synthesis of crystalline AuxAg1−x NP alloys in solution under ambient conditions, with the effective control over the size and the composition of the NPs possible by variation of the reaction time. Alloyed noble metal nanoparticles such as these are promising as sensor components due to their tunable optical extinction. The ability to easily produce crystalline bimetallic alloyed NPs under favourable conditions with real-time monitoring will afford a greater degree of control over the NP characteristics, and hence a superior product for implementation in a range of technological devices with precise specifications.
XXZ was supported by National Natural Science Foundation of China (Grants 11275127 and 90923005).
Notes and references
- Y. Chen, H. Cao, W. Shi, H. Liu and Y. Huang, Chem. Commun., 2013, 49, 5013 RSC.
- K. Shin, D. H. Kim and H. M. Lee, ChemSusChem, 2013, 6, 1044 CrossRef CAS PubMed.
- M. Sankar, N. Dimitratos, P. J. Miedziak, P. P. Wells, C. J. Kiely and G. J. Hutchings, Chem. Soc. Rev., 2012, 41, 8099 RSC.
- Q. Liu, Z. Zhao, Y. Lin, P. Guo, S. Li, D. Pan and X. Ji, Chem. Commun., 2011, 47, 964 RSC.
- P. N. Njoki, L. V. Solomon, W. Wu, R. Alama and M. M. Maye, Chem. Commun., 2011, 47, 10079 RSC.
- X. W. Liu and X. G. Liu, Angew. Chem., Int. Ed., 2012, 51, 3311 CrossRef CAS PubMed.
- A. K. Thapa, T. H. Shin, S. Ida, G. U. Sumanasekera, M. K. Sunkara and T. Ishihara, J. Power Sources, 2012, 220, 211 CrossRef CAS PubMed.
- R. E. Bailey and S. Nie, J. Am. Chem. Soc., 2003, 125, 7100 CrossRef CAS PubMed.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845 CrossRef CAS PubMed.
- M. G. Blaber, M. D. Arnold and M. J. Ford, J. Phys.: Condens. Matter, 2010, 22, 143201 CrossRef CAS PubMed.
- M. Armbrüster and Y. Grin, et al. , Nat. Mater., 2012, 11, 690 CrossRef PubMed.
- W.-H. Chiang and R. M. Sankaran, Nat. Mater., 2009, 8, 882 CrossRef CAS PubMed.
- W. Chen, R. Yu, L. Li, A. Wang, Q. Peng and Y. Li, Angew. Chem., Int. Ed., 2010, 49, 2917 CrossRef CAS PubMed.
- M. S. Shore, J. Wang, A. C. Johnston-Peck, A. L. Oldenburg and J. B. Tracy, Small, 2011, 7, 230 CrossRef CAS PubMed.
- M. P. Mallin and C. J. Murphy, Nano Lett., 2002, 2, 1235 CrossRef CAS.
- Q. Zhang, J.-Y. Lee, J. Yang, C. Boothroyd and J. Zhang, Nanotechnology, 2007, 18, 245605 CrossRef.
- N. Harris, M. J. Ford, P. Mulvaney and M. B. Cortie, Gold Bull., 2008, 41/1, 5 CrossRef.
- M. B. Cortie, X. Xu and M. J. Ford, Phys. Chem. Chem. Phys., 2006, 8, 3520 RSC.
- J. Wilcoxon, J. Phys. Chem. B, 2009, 113, 2647 CrossRef CAS.
- T. Udayabhaskararoo, Y. Sun, N. Goswami, S. K. Pal, K. Balasubramanian and T. Pradeep, Angew. Chem., Int. Ed., 2012, 51, 2155 CrossRef PubMed.
- A.-Q. Wang, J.-H. Liu, S. D. Lin, T.-S. Lin and C.-Y. Mou, J. Catal., 2005, 233, 186 CrossRef CAS PubMed.
- B. Xia, F. He and L. Li, Langmuir, 2013, 29, 4901 CrossRef CAS PubMed.
- X. Ren, X. Meng and F. F. Tang, Sens. Actuators, B, 2005, 110, 358 CrossRef CAS PubMed.
- J. Zheng, R. Yang, L. Xie, J. Qu, Y. Liu and X. Li, Adv. Mater., 2010, 22, 1451 CrossRef CAS PubMed.
- E. C. Neyts, A. C. T. van Duin and A. Bogaerts, J. Am. Chem. Soc., 2012, 134, 1256 CrossRef CAS PubMed.
- K. Ostrikov, E. C. Neyts and M. Meyyappan, Adv. Phys., 2013, 62, 113 CrossRef CAS.
- D. Mariotti and R. M. Sankaran, J. Phys. D: Appl. Phys., 2010, 43, 323001 CrossRef.
- X. Z. Huang, X. X. Zhong, Y. Lu, Y. S. Li, A. E. Rider, S. A. Furman and K. Ostrikov, Nanotechnology, 2013, 24, 095604 CrossRef CAS PubMed.
- C. Richmonds, M. Witzke, B. Bartling, S. W. Lee, J. Wainright, C. Liu and R. M. Sankaran, J. Am. Chem. Soc., 2011, 133, 17582 CrossRef CAS PubMed.
- J. Patel, L. Němcová, P. Maguire, W. G. Graham and D. Mariotti, Nanotechnology, 2013, 24, 245604 CrossRef CAS PubMed.
- D. Mariotti, J. Patel, V. Svrcek and P. Maguire, Plasma Processes Polym., 2012, 9, 1074 CrossRef CAS.
- M. Mozetic, U. Cvelbar, M. K. Sunkara and S. Vaddiraju, Adv. Mater., 2005, 17, 2138 CrossRef CAS.
- M. G. Kong, M. Keidar and K. Ostrikov, J. Phys. D: Appl. Phys., 2011, 44, 174018 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, energy efficiency estimates, spectroscopic and microanalysis results, other alloy nanoparticles produced, discussion of plasma effects, and comparison with other methods. See DOI: 10.1039/c3cc48846b |
| This journal is © The Royal Society of Chemistry 2014 |