 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect N9-arylation of purines with aryl halides†
Anders Foller
Larsen
and
Trond
Ulven
*
Department of Physics, Chemistry and Pharmacy, University of Southern Denmark, Campusvej 55, DK-5230 Odense M, Denmark. E-mail: ulven@sdu.dk
First published on 12th February 2014
Abstract
An efficient method for N-arylation of purines is reported. The N-arylation is catalysed by Cu(I) and 4,7-bis(2-hydroxyethylamino)-1,10-phenanthroline (BHPhen) in aqueous DMF or ethanol. The reaction generally proceeds with high selectivity for the N9-position.
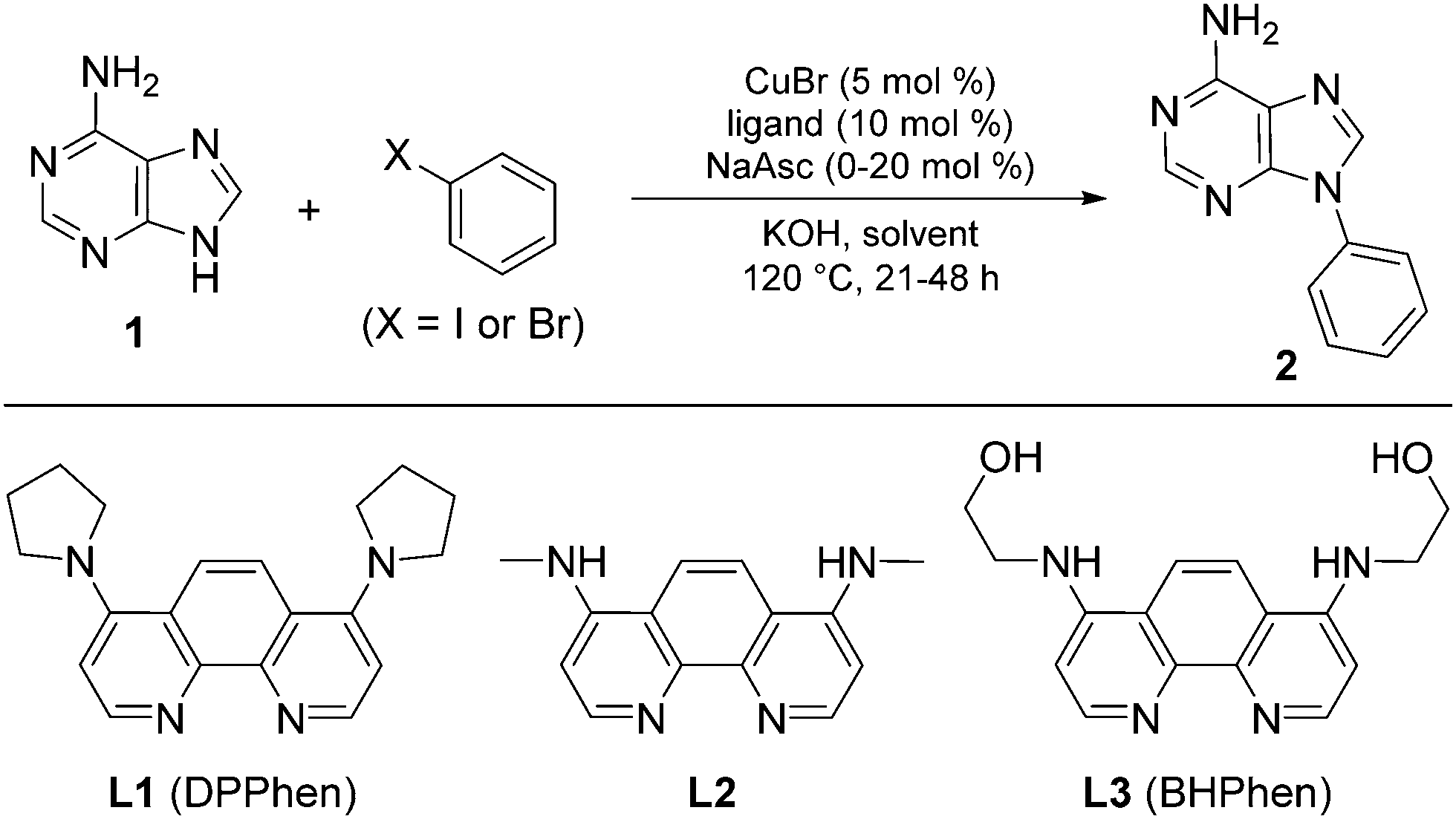
Purine is a ubiquitous heterocyclic scaffold of fundamental importance, and is considered to be a privileged structure in medicinal chemistry.1 Purine-based compounds, including N9-aryl purines, exhibit a wide range of biological and pharmaceutical activities,2 such as antiviral,3 antibacterial4 and anticancer5 effects. In light of their importance, there is a surprising paucity of efficient methods for N-arylation of purines. Despite the many methods for metal-catalysed C–N bond formation,6 only few examples involving purines are reported. Arylboronic acids and diaryliodonium salts have been used to achieve a Cu-mediated regioselective N9-arylation or N7-arylation of various purine derivatives in moderate to very good yields under relatively mild conditions,7 but these reactions often have a narrow substrate scope and N9-arylguanines have only been prepared by the arylation of protected or masked nucleobases.7b,d Aryl halides are abundantly available and are generally more stable than boronic acids and diaryliodonium salts; however, only three publications describe the N-arylation of purines with aryl halides, all in moderate yields.2b,8 Based on the hypothesis that electron-rich 1,10-phenanthroline ligands improve the performance by better stabilisation of the proposed Cu(III)-intermediate in the catalytic cycle, we recently found that 4,7-dipyrrolidinyl-1,10-phenanthroline (L1, DPPhen) catalyse N-arylation of various N-heterocycles with aryl halides in water. This system was also able to N-arylate adenine, 2,6-diaminopurine and theophylline, albeit only in moderate yields.9 Herein, we report a more general method for the direct synthesis of N-arylated purines with high and predictable selectivity for the N9- or N7-position over other potential coupling positions and complete selectivity over other nucleophilic sites including aromatic amines. The method is exemplified with various purines and a broad selection of aryl halides. Direct N-arylation of guanine and hypoxanthine are reported for the first time.
The coupling of adenine (1) with iodobenzene was selected for optimisation, initially using CuBr and ligand L1 with KOH as base in EtOH/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 120 °C (Table 1). EtOH/H2O was chosen since it readily produces a homogeneous mixture at this temperature. These conditions exclusively afforded the N9-arylated isomer in 46% yield with no trace of coupling at the N7- or 6-amino positions (entry 1). This complete regioselectivity is in accordance with selectivity observed in copper catalysed N-arylation of annelated imidazoles.10 Speculating that the moderate yield might be caused by in situ oxidation of Cu(I) to Cu(II), sodium ascorbate was added to the reaction mixture. This indeed improved the yield, with a 2:1 molar ratio between sodium ascorbate and copper giving the best results (entries 2–4). A ratio of 1.5:1 between iodobenzene and adenine provided the desired product in 83% yield (entry 5). Less polar reaction media, such as 1-butanol and water or pure ethanol, did not result in improved performance (entries 6 and 7), and more polar systems led to solubility problems.
:1) at 120 °C (Table 1). EtOH/H2O was chosen since it readily produces a homogeneous mixture at this temperature. These conditions exclusively afforded the N9-arylated isomer in 46% yield with no trace of coupling at the N7- or 6-amino positions (entry 1). This complete regioselectivity is in accordance with selectivity observed in copper catalysed N-arylation of annelated imidazoles.10 Speculating that the moderate yield might be caused by in situ oxidation of Cu(I) to Cu(II), sodium ascorbate was added to the reaction mixture. This indeed improved the yield, with a 2:1 molar ratio between sodium ascorbate and copper giving the best results (entries 2–4). A ratio of 1.5:1 between iodobenzene and adenine provided the desired product in 83% yield (entry 5). Less polar reaction media, such as 1-butanol and water or pure ethanol, did not result in improved performance (entries 6 and 7), and more polar systems led to solubility problems.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | X | Solventb | NaAsc (mol%) | Ratio PhX/1 | Yieldc (%) |
|
a Reaction conditions: 1 (0.50–0.75 mmol), aryl halide (PhX, 0.50–1.00 mmol), KOH (1.0 mmol), CuBr (5 mol%), ligand L1–L3 (10 mol%), sodium ascorbate (NaAsc, 0–20 mol%), solvent (1 mL), 120 °C, 21 h (X = I) or 48 h (X = Br).
b Solvent ratio was 4:1 (v/v).
c Yield of isolated product.
d 7.5 mol% L3.
e 15 mol% L3.
|
||||||
| 1 | L1 | I | EtOH–H2O | — | 1:1.2 |
46 |
| 2 | L1 | I | EtOH–H2O | 5 | 1:1.2 |
40 |
| 3 | L1 | I | EtOH–H2O | 10 | 1:1.2 |
70 |
| 4 | L1 | I | EtOH–H2O | 20 | 1:1.2 |
58 |
| 5 | L1 | I | EtOH–H2O | 10 | 1.5:1 |
83 |
| 6 | L1 | I | BuOH–H2O | 10 | 1.5:1 |
79 |
| 7 | L1 | I | EtOH | 10 | 1.5:1 |
42 |
| 8 | L1 | Br | EtOH–H2O | 10 | 2:1 |
75 |
| 9 | None | I | EtOH–H2O | 10 | 2:1 |
5 |
| 10 | L2 | Br | EtOH–H2O | 10 | 2:1 |
77 |
| 11 | L3 | Br | EtOH–H2O | 10 | 2:1 |
91 |
| 12 | L3 | I | EtOH–H2O | 10 | 1.5:1 |
90 |
| 13 | L3 | I | EtOH–H2O | 10 | 1.5:1 |
78 |
| 14 | L3 | I | EtOH–H2O | 10 | 1.5:1 |
64 |
| 15 | L3 | Br | EtOH–H2O | 10 | 1.5:1 |
85 |
| 16 | L3 | I | DMSO–H2O | 10 | 1.5:1 |
84 |
| 17 | L3 | Br | DMSO–H2O | 10 | 1.5:1 |
86 |
| 18 | L3 | I | DMF–H2O | 10 | 1.5:1 |
90 |
| 19 | L3 | Br | DMF–H2O | 10 | 1.5:1 |
87 |
Coupling with two equivalents of bromobenzene also gave good yield (entry 8), but we felt that the reaction could be improved further by reducing the amount of aryl halide. The phenanthroline ligand is critical for the reaction, as only trace amounts of the coupling product are observed when it is removed (entry 9). Ligand screening indicated poor general performance of bipyridines but superior performance of 4,7-dimethylaminophenanthroline L2 compared to L1 (see Table S1, ESI†). This can be rationalised by avoidance of steric interactions between the 4,7-substituents and the central phenanthroline ring for L2 and thereby higher planarity and increased electron donating effect of the amine substituents. Inspired by this result, we designed and synthesised phenanthroline ligand L3 with 2-hydroxyethylamino substituents. Interestingly, while ligand L2 afforded a yield similar to L1 in the optimised protocol (entry 10), L3 represented a substantial improvement, providing the desired product in excellent yield (entry 11). This may be ascribed to further increased electron donation from the 2-hydroxyethylamino substituents because of intramolecular hydrogen bonds between the amino nitrogen atoms and the hydroxyl groups, and possibly also to improved solubility properties.
Changing the ratio between copper and ligand to 1:1.5 gave a moderate drop in yield (entry 13), whereas a 1:3 ratio gave a pronounced drop (entry 14). This is in accordance with previous observations and is probably due to sequestering of copper by the excess ligand.9 Variation of the solvent system showed that DMSO/H2O and DMF/H2O also afforded very good yields at an aryl halide/adenine ratio of 1.5:1 for both iodobenzene and bromobenzene (entries 12 and 15–19), indicating a high robustness of the reaction. Of these, DMF/H2O was chosen as the medium for further investigation of the scope of the reaction, as it consistently gave high yields, produced a lower vapour pressure than EtOH/H2O, and was more readily removed than DMSO. It is well known that DMF/H2O can decompose to form dimethylamine when heated under basic conditions.11 While some dimethylamine indeed was observed in the reaction mixture, it did not interfere with the reaction, further supporting the robustness of this method.
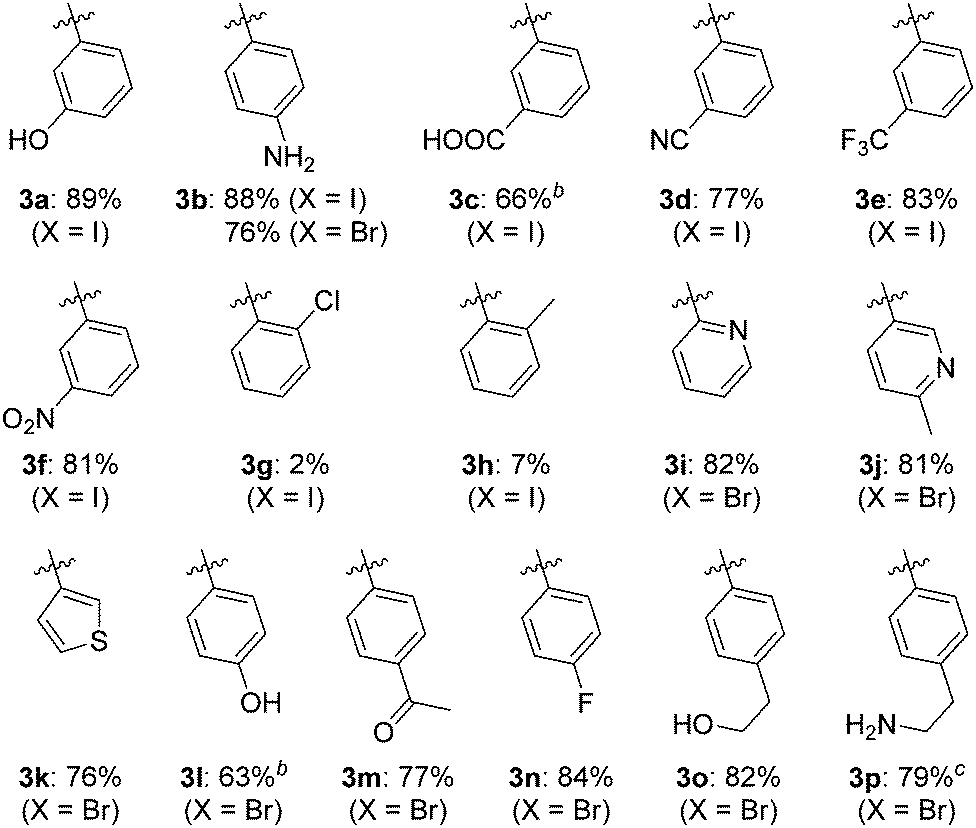
The substrate scope of the N-arylation was investigated by coupling adenine with aryl iodides and aryl bromides carrying various electron donating or withdrawing polar or non-polar substituents (Table 2). Alcohol, phenol, amine, carboxylic acid, cyano, trifluoromethyl, nitro, ketone and fluoro groups were all well tolerated and gave good to very good yields. However, substrates carrying substituents in the ortho position (3g and 3h) were poorly tolerated, probably because of unfavourable steric interactions with the Cu-ligand complex. 2-Bromopyridine, 5-bromo-2-methylpyridine and 3-bromothiophene coupled readily to yield 3i, 3j and 3k, respectively, indicating that heteroaryl halides are also suitable N-arylation substrates.12 When 4-bromophenethylamine was used as the substrate, formyl transfer from DMF was observed, resulting in isolation of the aliphatic formamide derivative of 3p as the only product in 79% yield. Switching to EtOH/H2O as reaction medium gave the expected primary amine 3p in 79% yield.
|
|
|---|
|
a Reaction conditions: 1 (0.50 mmol), aryl halide (0.75 mmol), KOH (1.0 mmol), CuBr (5 mol%), L3 (10 mol%), NaAsc (10 mol%), DMF/H2O (1 mL, 4:1 v/v), 120 °C, 21 h (X = I) or 48 h (X = Br). Yield of isolated product.
b Incomplete solubility of the reactant and product in the reaction medium.
c EtOH/H2O (1 mL, 4:1 v/v) was used as solvent. With DMF/H2O as solvent, the aliphatic N-formyl derivative of 3p was isolated in 79% yield.
|
|
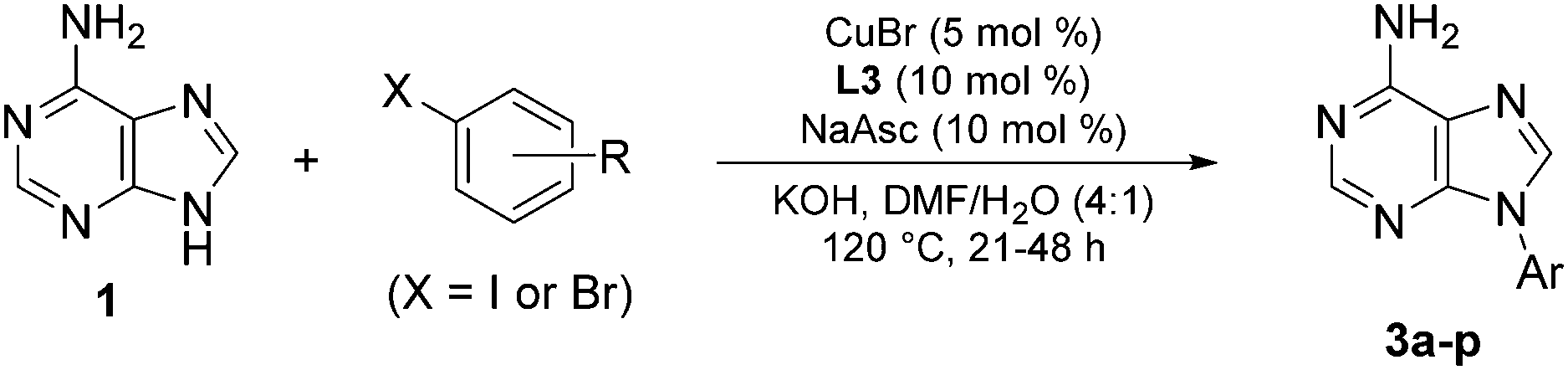
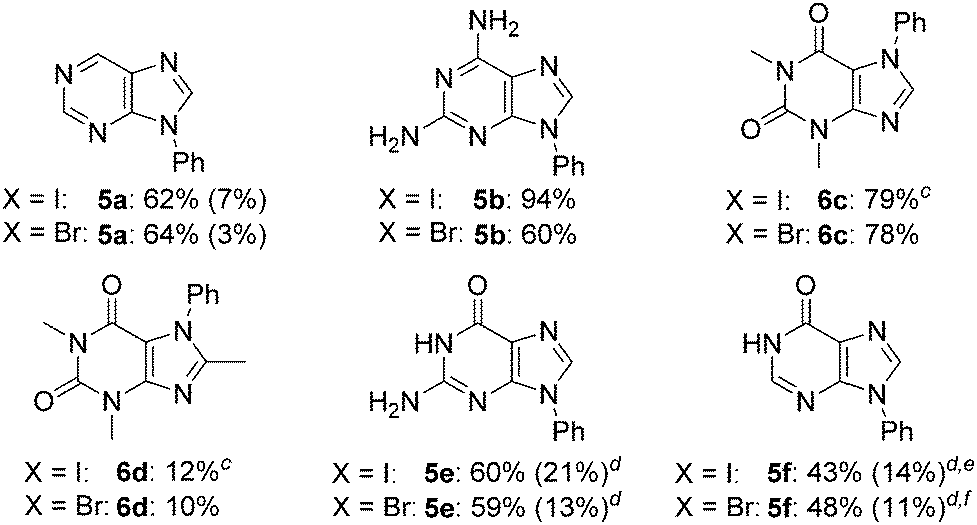
To further explore its scope, the protocol was investigated with various purine coupling partners (Table 3). Purine (4a) couples with iodo- and bromobenzene with a high selectivity for the N9- over the N7-position. This is rationalised by N3 exerting a directing effect on the catalytic complex. Coupling of 2,6-diaminopurine (4b) with iodo- and bromobenzene gave 94% and 60% yields, respectively, but with complete selectivity for the N9-position over the three other potential coupling positions in the molecule (N7 and the two amino groups). The exclusive selectivity for the N9- over the N7-position in both adenine and 2,6-diaminopurine indicates that the 6-amino group blocks N7-arylation. In contrast, theophylline (4c) couples with iodo- and bromobenzene to give 79% and 78% yields, respectively, of the exclusively N7-arylated product, which can be rationalised by steric hindrance of the N9-position by the N3-methyl substituent. The reaction of 8-methyltheophylline (4d) with iodo- and bromobenzene results in the isolation of the N7-arylated product in only 10–12% yield, indicating detrimental steric interactions between the C8-methyl and the Cu-ligand complex.
|
|
|---|
|
a Reaction conditions: 4a–f (0.50 mmol), aryl halide (PhX, 0.75 mmol), KOH (1.0 mmol), CuBr (5 mol%), L3 (10 mol%), NaAsc (10 mol%), DMF/H2O (1 mL, 4:1 v/v), 120 °C, 21 h (X = I) or 48 h (X = Br).
b Isolated yields, yields of the other (N7) regioisomers are given in parentheses.
c
t = 48 h.
d No added NaAsc; the solvent was DMSO/H2O (1 mL, 4:1 v/v).
e
t = 6 h.
f
t = 32 h.
|
|
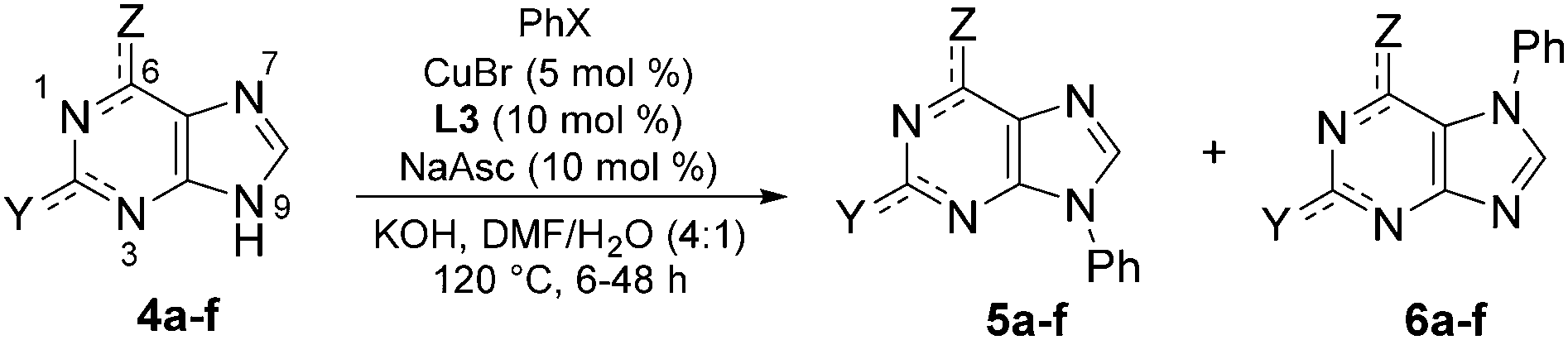
Guanine (4e) and hypoxanthine (4f) both couple with iodo- and bromobenzene with a lower preference for the N9-position over the N7-position compared to purine, reflecting a directing effect on the N7-position by the adjacent carbonyl group. The preference for the N9-position was enhanced by changing the solvent to DMSO/H2O and omitting sodium ascorbate. For both purines, this resulted in a ratio of isolated N9/N7-arylated product of 3:1 with iodobenzene and 9:2 with bromobenzene. In the case of hypoxanthine, arylation at N1 in addition to N9 was seen, and the reaction time was shortened to prevent over-arylation, resulting in somewhat lower yields compared to guanine, where no diarylated products were observed.
In conclusion, we have identified N4,N7-bis(2-hydroxyethyl)-1,10-phenanthroline-4,7-diamine (L3, BHPhen) as an enabling ligand for selective, direct copper-catalysed arylation of various common purines with aryl iodides and aryl bromides in moderate to excellent yields. Direct coupling of guanine and hypoxanthine with aryl halides were achieved for the first time. This is also the first report of direct coupling of purines with aryl bromides. Coupling of purines with aryl chlorides and sterically hindered aryl halides remains a challenge. A notable aspect is the predictable and generally high selectivity observed for arylation of one specific position in the presence of several other potential alkylation positions. Arylation at the N9-position was generally preferred, but this preference was altered in a predictable manner by the presence of adjacent groups. The robustness of the reaction is demonstrated not only by the broad substrate scope, but also by a good performance in several different solvent systems. Although DMF/H2O was chosen as the standard for the reactions reported herein, the EtOH/H2O system might be more suitable for up-scaling, where the cost and environmental impact become important factors.
We are grateful for financial support from the Danish Council for Independent Research, Technology and Production, Grants 274-08-056 and 09-070364.
Notes and references
- (a) H. Rosemeyer, Chem. Biodiversity, 2004, 1, 361 CrossRef CAS PubMed; (b) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347 CrossRef CAS PubMed.
- (a) M. Legraverend and D. S. Grierson, Bioorg. Med. Chem., 2006, 14, 3987 CrossRef CAS PubMed; (b) N. Kato, T. Sakata, G. Breton, K. G. Le Roch, A. Nagle, C. Andersen, B. Bursulaya, K. Henson, J. Johnson, K. A. Kumar, F. Marr, D. Mason, C. McNamara, D. Plouffe, V. Ramachandran, M. Spooner, T. Tuntland, Y. Zhou, E. C. Peters, A. Chatterjee, P. G. Schultz, G. E. Ward, N. Gray, J. Harper and E. A. Winzeler, Nat. Chem. Biol., 2008, 4, 347 CrossRef CAS PubMed; (c) F. Di Virgilio, Cancer Res., 2012, 72, 5441 CrossRef CAS PubMed.
- L. Aguado, H. J. Thibaut, E.-M. Priego, M.-L. Jimeno, M.-J. Camarasa, J. Neyts and M.-J. Pérez-Pérez, J. Med. Chem., 2009, 53, 316 CrossRef PubMed.
- A. K. Bakkestuen, L.-L. Gundersen and B. T. Utenova, J. Med. Chem., 2005, 48, 2710 CrossRef CAS PubMed.
- N. Kode, L. Chen, D. Murthy, D. Adewumi and S. Phadtare, Eur. J. Med. Chem., 2007, 42, 327 CrossRef CAS PubMed.
- (a) I. P. Beletskaya and A. V. Cheprakov, Organometallics, 2012, 31, 7753 CrossRef CAS; (b) T. R. M. Rauws and B. U. W. Maes, Chem. Soc. Rev., 2012, 41, 2463 RSC; (c) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed.
- (a) A. K. Bakkestuen and L.-L. Gundersen, Tetrahedron Lett., 2003, 44, 3359 CrossRef CAS; (b) M. F. Jacobsen, M. M. Knudsen and K. V. Gothelf, J. Org. Chem., 2006, 71, 9183 CrossRef CAS PubMed; (c) L. Tao, Y. Yue, J. Zhang, S.-Y. Chen and X.-Q. Yu, Helv. Chim. Acta, 2008, 91, 1008 CrossRef CAS; (d) R. Keder, H. Dvořáková and D. Dvořák, Eur. J. Org. Chem., 2009, 1522 CrossRef CAS; (e) H.-Y. Niu, C. Xia, G.-R. Qu, Q. Zhang, Y. Jiang, R.-Z. Mao, D.-Y. Li and H.-M. Guo, Org. Biomol. Chem., 2011, 9, 5039 RSC; (f) I. Čerňa, R. Pohl, B. Klepetářová and M. Hocek, J. Org. Chem., 2008, 73, 9048 CrossRef PubMed; (g) S. Ding, N. S. Gray, Q. Ding and P. G. Schultz, Tetrahedron Lett., 2001, 42, 8751 CrossRef CAS; (h) E. Kiselgof, D. B. Tulshian, L. Arik, H. Zhang and A. Fawzi, Bioorg. Med. Chem. Lett., 2005, 15, 2119 CrossRef CAS PubMed; (i) I. Dreier, S. Kumar, H. Søndergaard, M. L. Rasmussen, L. H. Hansen, N. H. List, J. Kongsted, B. Vester and P. Nielsen, J. Med. Chem., 2012, 55, 2067 CrossRef CAS PubMed.
- (a) J. C. Antilla, J. M. Baskin, T. E. Barder and S. L. Buchwald, J. Org. Chem., 2004, 69, 5578 CrossRef CAS PubMed; (b) M. N. Soltani Rad, S. Behrouz, M. M. Doroodmand and N. Moghtaderi, Synthesis, 2011, 3915 CrossRef PubMed.
- J. Engel-Andreasen, B. Shimpukade and T. Ulven, Green Chem., 2013, 15, 336 RSC.
- (a) S. Ueda and S. L. Buchwald, Angew. Chem., Int. Ed., 2012, 51, 10364 CrossRef CAS PubMed; (b) N. T. Jui and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 11624 CrossRef CAS PubMed.
- (a) A. Agarwal and P. M. S. Chauhan, Synth. Commun., 2004, 34, 2925 CrossRef CAS PubMed; (b) L. Čechová, P. Jansa, M. Šála, M. Dračínský, A. Holý and Z. Janeba, Tetrahedron, 2011, 67, 866 CrossRef PubMed; (c) T. P. Petersen, A. F. Larsen, A. Ritzén and T. Ulven, J. Org. Chem., 2013, 78, 4190 CrossRef CAS PubMed.
- A control reaction without CuBr gave only 7% conversion after 48 h, indicating that nucleophilic aromatic substitution only makes a minor contribution to the yield of 3i.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and full spectroscopic data for all new compounds. See DOI: 10.1039/c3cc48642g |
| This journal is © The Royal Society of Chemistry 2014 |