 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A chemical “hub” for absolute quantification of a targeted protein: orthogonal integration of elemental and molecular mass spectrometry†
Xiaowen
Yan
a,
Zhaoxin
Li
a,
Yong
Liang
a,
Limin
Yang
a,
Bo
Zhang
a and
Qiuquan
Wang
*ab
aDepartment of Chemistry and the MOE Key Laboratory of Spectrochemical Analysis & Instrumentation, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: qqwang@xmu.edu.cn; Fax: +86(0)599-2187400
bState Key Laboratory of Marine Environmental Science, Xiamen University, Xiamen 361005, China
First published on 5th May 2014
Abstract
A novel element-tagged activity-based photo-cleavable biotinylated chemical “Hub” was designed and synthesized to orthogonally integrate ICP-MS and ESI-MS for absolute targeted-protein quantification. This tetrafunctional chemical “hub” allowed us to quantify a targeted protein using species-unspecific isotope dilution ICP-MS and to know which protein is being quantified using ESI-IT-MS at the same time.
After decades of development, ESI- and/or MALDI-based molecular mass spectrometry (ESI-/MALDI-MS) with isotope-labeling strategies is widely used as a relative protein quantification tool in addition to its powerful structure-identification ability.1 Together with the use of stable isotope-labeled proteotypic peptides as internal standards, ESI-MS with its triple-quadrupole mass analyzer enables the absolute quantification of proteins through multiple reaction monitoring (MRM) of the proteotypic peptides.2 However, a high-quality MRM quantification calls for the synthesis of the isotope-labeled proteotypic peptide standards for the targeted proteins, whose sequences and properties must be well-known. This implies that even known proteins are difficult to be quantified with this latter technique if the proteotypic peptide standards could not be accurately synthesized. Moreover, it is also extremely difficult and expensive to synthesize the numerous isotope-labeled proteotypic peptide standards for the hundreds and thousands of proteins in a complex proteome.3
Absolute protein quantification, as one of the most important research directions of proteomics, can precisely number the protein copies in a cell or tissue sample, and thus describes the role of the protein in a complex molecular network of life. It signifies not only the exact content of a protein but also comprehensive molecular information concerning the protein to be quantified. Compared with the ESI-/MALDI-MS, elemental mass spectrometry, especially the very hard ionization source based inductively coupled plasma mass spectrometry (ICP-MS), has been recently proved to be an attractive technique for absolute protein quantification using only one element/isotope standard due to its unique feature that the signal of an element/isotope naturally in and/or tagged on a protein is independent of the kind and chemical form of the protein. This means that any known species of the element/isotope can be used as a standard for absolute protein quantification using species-unspecific isotope dilution (SUID) ICP-MS.4 Unfortunately, ICP-MS is unable to provide any structural information about the protein to be quantified. Because the protein might have and/or be tagged by an identical element/isotope, not only mutual separation of the targeted one from coexisting proteins but also the stoichiometry of the element/isotope in/on the protein must be known before ICP-MS quantification. In this case, the powerful ESI-/MALDI-MS should be employed working together with ICP-MS to release the “one standard protein quantification” advantage of ICP-MS, and especially to know what protein is being quantified. In this regard, we need a chemical “hub” to orthogonally integrate ICP-MS and ESI/MALDI-MS rather than physically using them in parallel for achieving absolute targeted-protein quantification and identification.
In this proof-of-concept study, we report the design and synthesis of a novel activity-based element-tagged photo-cleavable biotinylated chemical “hub” to orthogonally integrate ICP-MS and ESI ion trap MS (ESI-IT-MS) for the first time. In general, the tetrafunctional chemical “hub” as shown in Scheme 1 comprises the following four parts: (1) a sulfonyl fluoride as an example of guiding groups, which has specificity towards the hydroxyl group of the serine residue at the active site of serine proteases (SPs); (2) a Eu-loaded DOTA to produce ICP-MS signals for protein quantification; (3) a conjugated biotin for being captured by streptavidin-coated beads; and (4) a photo-cleavable o-nitrobenzyl ether linker to release the captured-SP upon UV irradiation for ESI-IT-MS identification. To build all these parts into the tetrafunctional chemical “hub”, we designed a synthetic route as illustrated in Fig. 1 (see details in the ESI†). Briefly, 4-[4-(1-hydroxyethyl)-2-methoxy-5-nitrophenoxy] butyric acid (1) was first conjugated with N-Fmoc-1,4-butanediamine (2) to produce compound 3, the hydroxyl group of which was activated with N,N′-disuccinimidyl carbonate (4) to obtain the photocleavable linker Fmoc-nitrobenzyl-NHS (5). To synthesize the Eu-chelating moiety amine-DOTA-azide (10), N-Boc-6-azido-L-norleucine (6) was first conjugated with 2 to produce compound 7, whose amino group was deprotected by removing the Boc group in TFA and reacted with DOTA-NHS (9) to give 10. Next, compound 5 was conjugated with 10 to produce Fmoc-nitrobenzyl-DOTA-azide (11). The Fmoc was subsequently removed from the amine group of compound 11 and reacted with (+)-Biotin-PEG4-NHS to obtain biotin-nitrobenzyl-DOTA-azide (12). The Eu3+-loaded 12 was finally conjugated with compound 13 through the Copper-Catalyzed Azide–Alkyne Cycloaddition (CuAAC) reaction5 to obtain the tetrafunctional chemical “hub” 14, biotin-PEG-nitrobenzyl-DOTA-Eu-SF. It was stable not only during its use but also up to 6 months (longer time was not tested) if we dissolved it in anhydrous DMSO and kept in the dark at −20 °C. The details of the synthetic processes, purifying method and the related data including HPLC-UV/MS, 1H-NMR and 13C-NMR are all described in the ESI† (Fig. S1–S18).
 | ||
| Scheme 1 Chemical structure of the tetrafunctional chemical “hub” and procedures for applying it for specifically tagging and capturing as well as releasing the targeted protease for quantification and identification using ICP-MS and ESI-MS. | ||
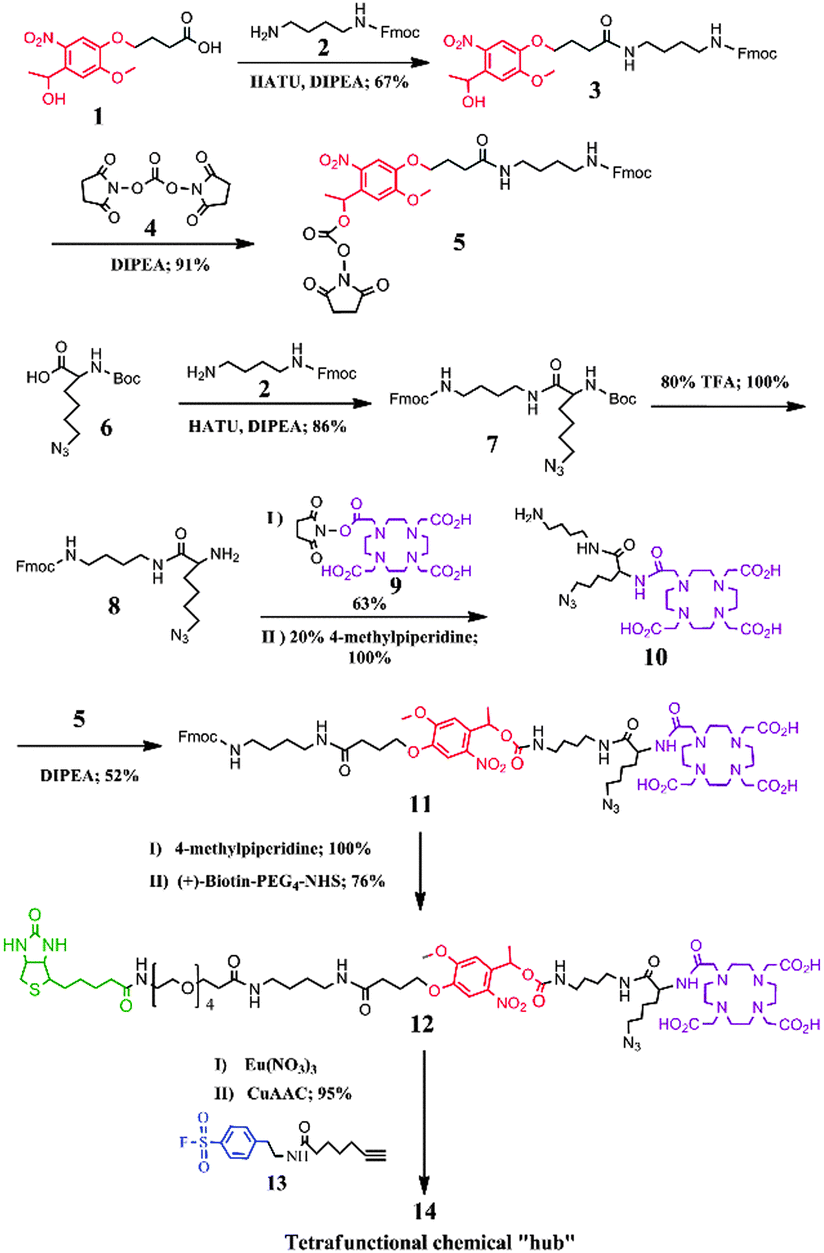 | ||
| Fig. 1 Synthetic route of tetrafunctional chemical “hub” 14. Details of the synthetic procedures are presented in the ESI.† | ||
In order to investigate the cleavage efficiency of the “hub” 14, 1 mM 14 in 100 mM Tris-HCl (pH 7.5) was analyzed using HPLC-UV/ESI-IT-MS without and with the irradiation of UV light (λ = 365 nm) for 20 min. The results obtained indicated that the intact 14 was eluted at 28 min on a C18 column (1.0 I.D. × 15.0 mm in length) (Fig. 2a) under the optimized separation conditions (see the ESI†). The deconvolution molecular weight (DM) of 14 was determined to be 1957 Da using ESI-IT-MS (Fig. 2c), identical to its theoretical molecular weight (TM, 1957 Da); and neither cleavage product nor forfeiture of Eu3+ was observed, indicating that 14 was stable during chromatographic separation. From the HPLC chromatogram shown in Fig. 2b, on the other hand, we could see that almost all of 14 disappeared after 20 min of UV irradiation, and the cleavage efficiency was calculated to be 99% by comparing the peak areas of 14 before and after UV irradiation. One cleaved product of 14 was found to be eluted at a retention time of 23 min (Fig. 2b), and was assigned to be the SF-DOTA-Eu moiety (TM = 1088 Da) (Fig. 2d), which is consistent with the photocleavage mechanism of the o-nitrobenzyl ether compound (Fig. S19 in the ESI†).6 We did not find the other part of UV-cleaved 14. The reason was speculated to be the low ionization efficiency of this biotin-containing part in ESI-IT-MS. In any event, 14 was proven to undergo swift photolysis under UV irradiation.
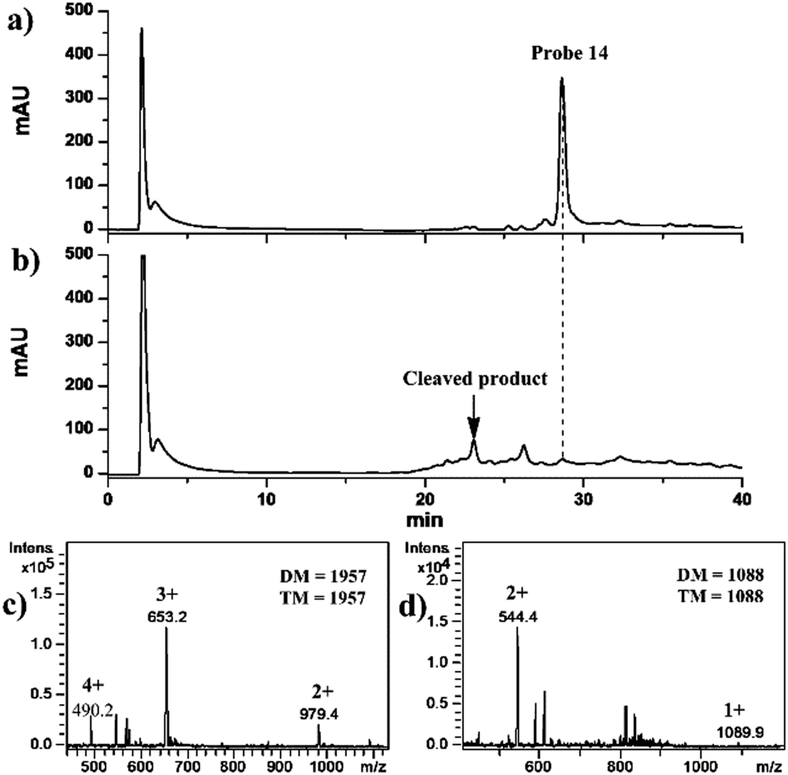 | ||
| Fig. 2 HPLC-UV (214 nm) chromatograms (a and b) of the tetrafunctional chemical “hub” 14 and its mass spectra (c and d) before and after UV irradiation. DM and TM in the mass spectra denote devolution and theoretical molecular weights. | ||
The tagging efficacy of 14 towards a typical SP, bovine chymotrypsin with a DM value of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 438 Da (Fig. S20a in ESI†) that was selected as an example in this proof-of-concept study, was then investigated using ESI-IT-MS. After incubation of 1 mM 14 with freshly prepared chymotrypsin in 100 mM Tris-HCl (pH 7.5) for 1 hour, the DM value of the tagged chymotrypsin was found to be 27376 Da, matching the TM value of 27375 [25438 (chymotrypsin) + 1957 (14) – 20 (HF)] Da well (Fig. S20b in ESI†), suggesting that one chymotrypsin molecule was tagged with one 14. The difference of 1 Da between the DM and TM values was due to the relative low-mass resolution of the ESI-IT-MS used in this study. This result proved that the sulfonyl fluoride group in 14 has high specificity to the hydroxyl group of the serine residue at the active site of chymotrypsin,7 even though there are numbers of serine residues and hydroxyl groups in other parts of this protein, demonstrating an excellent intra-hydroxyl group selectivity.
438 Da (Fig. S20a in ESI†) that was selected as an example in this proof-of-concept study, was then investigated using ESI-IT-MS. After incubation of 1 mM 14 with freshly prepared chymotrypsin in 100 mM Tris-HCl (pH 7.5) for 1 hour, the DM value of the tagged chymotrypsin was found to be 27376 Da, matching the TM value of 27375 [25438 (chymotrypsin) + 1957 (14) – 20 (HF)] Da well (Fig. S20b in ESI†), suggesting that one chymotrypsin molecule was tagged with one 14. The difference of 1 Da between the DM and TM values was due to the relative low-mass resolution of the ESI-IT-MS used in this study. This result proved that the sulfonyl fluoride group in 14 has high specificity to the hydroxyl group of the serine residue at the active site of chymotrypsin,7 even though there are numbers of serine residues and hydroxyl groups in other parts of this protein, demonstrating an excellent intra-hydroxyl group selectivity.
Subsequently, a sample containing seven non-SP proteins (RNase A, insulin, Cyt c, lysozyme, BSA, carbonic anhydrase and ovalbumin) and one SP (chymotrypsin) was chosen to further evaluate the inter-protein specificity of 14 (Table S1 in the ESI†) via HPLC coupled with UV detection (214 nm) and 153Eu-SUID ICP-MS. As shown in Fig. 3a, the eight proteins were separated on a C18 column (see the details in the ESI†), whereas the retention time of 14 was the same as that of Cyt c coincidentally, and BSA partly overlapped with chymotrypsin under the HPLC conditions employed in this study. When the effluent from the C18 column was mixed on-line with the enriched 153Eu standard solution through a three-way connector, and directly analyzed by ICP-MS monitoring of the signals of 153Eu and 151Eu isotopes without protein capture and release steps, only chymotrypsin was determined (Fig. 3b) due to the element-specific feature of ICP-MS, suggesting that only chymotrypsin was tagged by 14 and demonstrating the activity-based inter-protein specificity of 14 towards chymotrypsin even though there are many serine residues in the other seven proteins. It should be pointed out that although BSA, which is a high abundant protein in biological samples, overlapped with chymotrypsin during the HPLC separation with UV detection, the interference was blinded when using ICP-MS. This is very important considering the limited separating ability of current available separation techniques that frequently encountered the problem to achieve a quantitative separation of the individual proteins in a complex biological sample. Besides, although the signal of 14 might overlap the possible signal of Cyt c in the sample, no signal was observed in the same retention time after 14 was UV-cleaved (data not shown here). It is worth mentioning that, furthermore, the signal intensity of Eu decreased along with the increase in the acetonitrile content in the HPLC mobile phase as shown in Fig. 3b, which suggested that the signal response of Eu is lower at higher retention time (higher acetonitrile content). This effect can be effectively calibrated by SUID, since the signal of 151Eu representing the tagged protein in the sample and that from the spiked 153Eu standard solution are affected to the same extent. The chromatograms of 151Eu and 153Eu isotopes were then transformed into the Eu mass-flow chromatogram (Fig. 3c) using the online isotope dilution equation.8 A limit of detection (LOD, 3σ) of 0.5 fmol chymotrypsin was obtained with RSD lower than 3% (n = 5 at pmol level). This LOD is about 40-fold higher than the sensitivity of conventional enzyme-linked immunosorbent assay (ELISA) (21 fmol), and comparable to the state-of-the-art single-molecule ELISA (0.4 fmol),9 besides the advantage that this chemical “hub” integrated ICP-MS and ESI-IT-MS offers both quantitative and structural information. The concentration of chymotrypsin in the mixed protein sample, prepared from the crude chymotrypsin solid powder obtained from Sigma-Aldrich, was determined to be (17.1 ± 1.0) μM by integrating the peak area of chymotrypsin as shown in Fig. 3c, which corresponded to the chymotrypsin content of (86.9 ± 5.1)% in the crude solid powder with the recovery of (91.3 ± 5.4)% according to the data provided by the producer.
 | ||
| Fig. 3 HPLC chromatograms of a 14-tagged protein sample determined using (a) UV (214 nm) and (b) 153Eu species-unspecific isotope dilution ICP-MS, (c) Eu mass flow chromatogram of the 14-tagged protein transformed from (b) based on the online isotope dilution equation, (d) the supernatant after the protein sample was labeled with 14 and captured using streptavidin-coated magnetic beads, (e) the supernatant determined using UV (214 nm) after the captured SF-DOTA-Eu tagged chymotrypsin was released after 20 minutes of UV (365 nm) irradiation, and (f) ESI-IT-MS of peak 6 in (e). 1, RNase A; 2, insulin; 3, Cyt c; 4, lysozyme; 5, BSA; 6, chymotrypsin; 7, carbonic anhydrase; 8, ovalbumin. | ||
Finally, in order to evaluate the fishing and releasing ability of the chemical “hub” 14 and to perform ESI-IT-MS identification to know what protein is being quantified by ICP-MS, the above protein sample was used again. The sample was incubated with 14, followed by fishing with streptavidin-coated magnetic beads and washing with Tris-HCl buffer (Scheme 1 and details in the ESI†). As shown in Fig. 3d, the seven proteins except chymotrypsin were observed in the chromatogram, demonstrating a very efficient fishing of chymotrypsin. After the magnetically separated beads were resuspended in Tris-HCl buffer and irradiated under UV light for 20 min, the supernatant obtained was analyzed again (Fig. 3e, peak 6), and the HPLC effluent was subsequently infused into ESI-IT-MS for identification (Fig. 3f). The DM value of the analyte in peak 6 was found to be 26507 Da, which is consistent with the TM value of 26506 Da of DOTA-Eu-SF-tagged-chymotrypsin, confirming that the protein in peak 6 was chymotrypsin. All these results demonstrated that 14 was an effective tetrafunctional chemical “hub” to orthogonally integrate ICP-MS and ESI-IT-MS, allowing us to know what has been quantified using ICP-MS. Following our previous study on quantification of ICP-MS based SPs in the Tris-HCl extracts of rat tissues using sulfonyl fluoride, the same as the activity-based targeting reagent used in this study,8 the obtained results indicated that SPs ranged 0.4 to 7.6 nmol g−1, and the recoveries (%) of the spiked chymotrypsin and elastase (another typical SP) were 91.3 and 90.5 (kidney), 96.0 and 89.6 (heart), 91.0 and 97.5 (liver), 93.3 and 90.4 (lung), and 95.2 and 90.1 (brain), as well as 38.4 and 96.7 (pancreas), demonstrating the efficacy of this chemical “hub” methodology. The lower recovery of chymotrypsin in pancreas is because the endogenous inhibitors in the pancreas extract significantly inhibited its activity.10
In summary, for the first time to our knowledge, we designed and synthesized a novel activity-based element-tagged photo-cleavable biotinylated chemical “hub” to orthogonally integrate ICP-MS and ESI-IT-MS in this proof-of-concept study. The chemical “hub” makes it possible to use these two powerful tools exerting their respective merits of absolute quantification and qualitative identification of a targeted protein at the same time. As a versatile approach, we have built up the frame of the chemical “hub” (compound 12 in this proof-of-concept study). The azide group in 12 offers the possibility of conjugating other biospecific guiding groups through the CuAAC reaction, and other lanthanide (and its isotope) ions can be easily coordinated into the DOTA moiety to construct the corresponding chemical “hub” towards other targeted proteins. In this way, the strategy based on the chemical “hub” can be easily extended to the quantification and identification of other targeted proteases such as cysteine, threonine and aspartic proteases.11 We believe that this chemical “hub” strategy sets an example, and will open the opportunity not only to acquire quantitative information of a targeted known protein but also to discover new members in a certain family of proteins, thus providing more and more comprehensive information concerning the proteins for further biological and medical studies in the near future.
This study was financially supported by the National Basic Research 973 Program (2014CB932004) and the National Natural Science Foundation of China (21035006 and 21275120). We thank Prof. John Hodgkiss of The University of Hong Kong for helping with the English in this article.
References
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS PubMed.
- (a) S. A. Gerber, J. Rush, O. Stemman, M. W. Kirschner and S. P. Gygi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6940–6945 CrossRef CAS PubMed; (b) L. Anderson and C. L. Hunter, Mol. Cell. Proteomics, 2006, 5, 573–588 CrossRef CAS PubMed.
- J. R. Whiteaker, C. Lin, J. Kennedy, L. Hou, M. Trute, I. Sokal, P. Yan, R. M. Schoenherr, L. Zhao, U. J. Voytovich, K. S. Kelly-Spratt, A. Krasnoselsky, P. R. Gafken, J. M. Hogan, L. A. Jones, P. Wang, L. Amon, L. A. Chodosh, P. S. Nelson, M. W. McIntosh, C. J. Kemp and A. G. Paulovich, Nat. Biotechnol., 2011, 29, 625–634 CrossRef CAS PubMed.
- (a) A. Sanz-Medel, M. Montes-Bayon, J. Bettmer, M. L. Fernandez-Sanchez and J. R. Encinar, Trends Anal. Chem., 2012, 40, 52–63 CrossRef CAS PubMed; (b) X. W. Yan, L. M. Yang and Q. Q. Wang, Anal. Bioanal. Chem., 2013, 405, 5663–5670 CrossRef CAS PubMed.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed; (c) T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS PubMed.
- (a) J. E. Corrie, A. Barth, V. R. Munasinghe, D. R. Trentham and M. C. Hutter, J. Am. Chem. Soc., 2003, 125, 8546–8554 CrossRef CAS PubMed; (b) Y. V. Il'ichev, M. A. Schwörer and J. Wirz, J. Am. Chem. Soc., 2004, 126, 4581–4595 CrossRef CAS PubMed.
- J. C. Powers, J. L. Asgian, O. D. Ekici and K. E. James, Chem. Rev., 2002, 102, 4639–4750 CrossRef CAS PubMed.
- (a) L. Rottmann and K. G. Heumann, Anal. Chem., 1994, 66, 3709–3715 CrossRef CAS; (b) J. Vogl and K. G. Heumann, Anal. Chem., 1998, 70, 2038–2043 CrossRef CAS; (c) X. W. Yan, Y. C. Luo, Z. B. Zhang, Z. X. Li, Q. Luo, L. M. Yang, B. Zhang, H. F. Chen, P. M. Bai and Q. Q. Wang, Angew. Chem., Int. Ed., 2012, 51, 3358–3363 CrossRef CAS PubMed.
- D. M. Rissin, C. W. Kan, T. G. Campbell, S. C. Howes, D. R. Fournier, L. Song, T. Piech, P. P. Patel, L. Chang, A. J. Rivnak, E. P. Ferrell, J. D. Randall, G. K. Provuncher, D. R. Walt and D. C. Duffy, Nat. Biotechnol., 2010, 28, 595–599 CrossRef CAS PubMed.
- Y. T. Wachtfogel, U. Kucich, C. E. Hack, P. Gluszko, S. Niewiarowski, R. W. Colman and L. H. Edmunds Jr., J. Thorac. Cardiovasc. Surg., 1993, 106, 1–9 CAS.
- C. López-Otín and L. M. Matrisian, Nat. Rev. Cancer, 2007, 7, 800–808 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supporting figures. See DOI: 10.1039/c3cc48460b |
| This journal is © The Royal Society of Chemistry 2014 |