 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new gold-catalysed azidation of allenes†
César
Hurtado-Rodrigo
,
Stefanie
Hoehne
and
María Paz
Muñoz
*
School of Chemistry, University of East Anglia, NR4 7TJ, Norwich, UK. E-mail: m.munoz-herranz@uea.ac.uk; Fax: +44 (0) 1603592003
First published on 2nd December 2013
Abstract
A new gold-catalysed azidation reaction of allenes is presented as a new highly modular approach for the synthesis of substituted allyl derivatives containing nitrogen from simple precursors.
Organic azides are common building blocks for the synthesis of natural products and nitrogen-containing heterocycles of pharmacological relevance.1 As classic azides, allyl azides are useful precursors of many functional groups (e.g. amines, nitriles),2 and can be used in copper-catalysed 1,3-dipolar additions to alkynes for the synthesis of allyl triazoles.3 The classic synthesis of allyl azides involves the substitution of primary or secondary allyl derivatives with nucleophilic azides, like NaN3 or TMSN3 (Scheme 1a).4 However these methods have limitations, and mixtures of regioisomers are often obtained. Pd- and Mo-catalysed allylic azidations have been also described with better results in the selectivity and scope.5
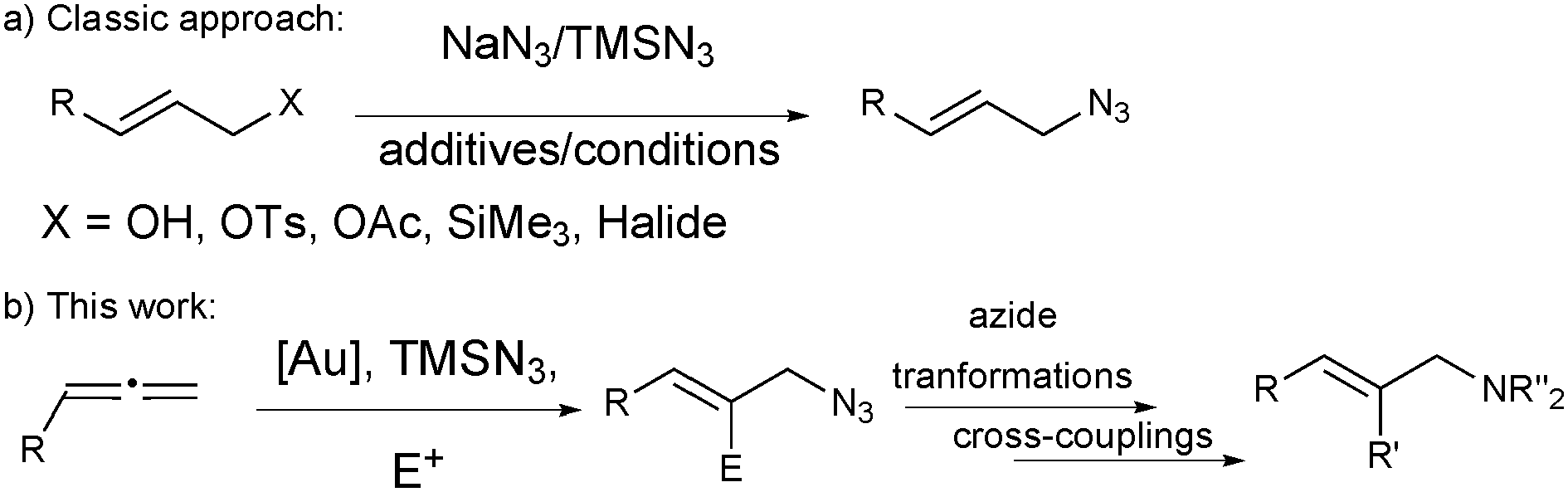 | ||
| Scheme 1 Different approaches for the synthesis of allyl azides. | ||
Allenes are versatile starting materials for developing sequential reactions, affording new and efficient methods for the preparation of complex compounds of synthetic or biological importance. In this area, gold-catalysed intermolecular addition of nucleophiles to allenes is an important reaction that has been widely developed in the last few years, especially for oxygen nucleophiles,6 although the nitrogen7 and carbon-based nucleophile8 versions have also been described. We envisioned that nucleophilic azides could be used in a similar gold-catalysed reaction9 with allenes,10 as a new method for the synthesis of allyl azides. We report here the first examples of this concept and our efforts to develop this new methodology as a highly modular approach for the synthesis of allyl azides with extra functionalities that will enable a straightforward increase in molecular complexity from simple allenes (Scheme 1b).
The challenge of this reaction is that, in contrast with alcohols or amines, the available nucleophilic azides, NaN3 or TMSN3, do not have the necessary proton to furnish the protonolysis of the vinyl–gold complex intermediate and therefore, the catalytic cycle cannot be completed.11 In order to close the catalytic cycle, the volatile, toxic and explosive hydrazoic acid, HN3,12 has to be cautiously generated in situ.13 Nevertheless, protonolysis of vinyl–gold(I) complexes is only one way to close the catalytic cycle and other electrophiles can be employed to break the C–Au bond without the need for hazardous reagents.14
Using commercially available cyclohexylallene (1a) and Ph3PAuCl/AgOTf, we first investigated the optimal conditions for the hydroazidation reaction. We found that TMSN3 was the best source of the nucleophilic azide, and trifluoroacetic acid (TFA) in DCM was the best proton source to provide clean complete conversion to the expected allyl azide 2a, with its regioisomer 2a′, together with allyl trifluoroacetate, 3a, plus acetamide 4a as an unexpected product (Scheme 2).15
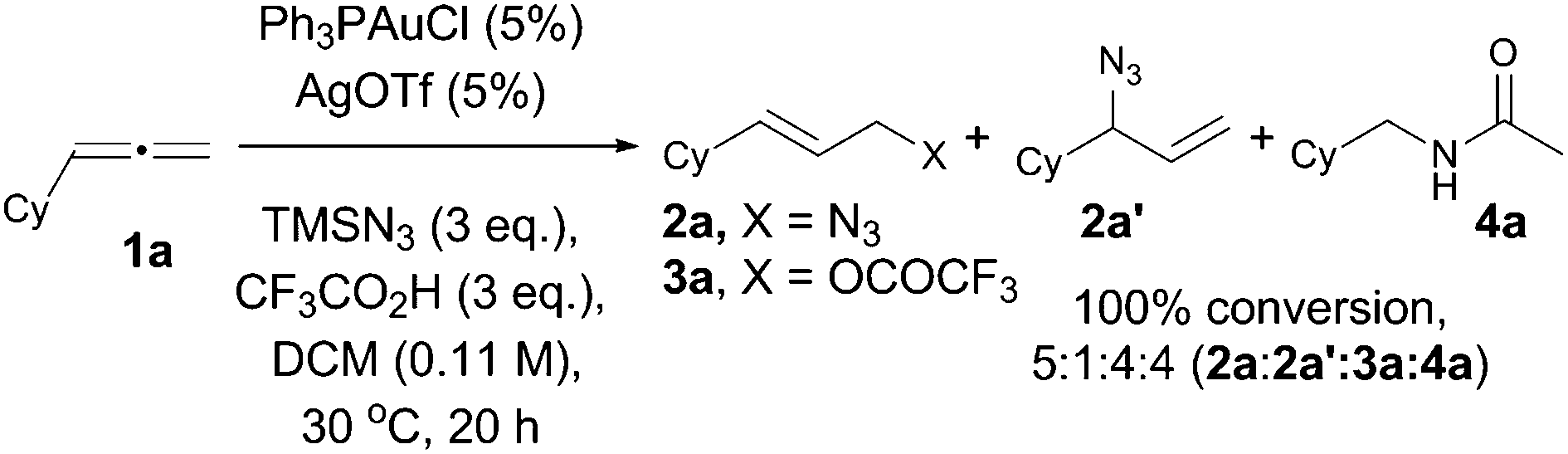 | ||
| Scheme 2 Gold-catalysed hydroazidation of cyclohexylallene. | ||
AgOTf seemed to be the most effective chloride abstractor in this reaction, and was used for catalyst screening.16 NHC–Au complexes did not favour the azidation reaction, and 3a was obtained as the major component in low conversions. The best result was obtained with (PhO)3PAuCl as the catalyst. In a final approach to get the optimum conditions, we found that the best ratio of products and yields was obtained changing the concentration, lowering the temperature, adding up to 5 equivalents of water as additive,17,18 and purifying the mixture over basic alumina. Under the best conditions, we investigated the scope of the reaction with different substituted allenes (Table 1).
|
|
||
|---|---|---|
| Entrya | Allene | Yield (%), 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2′ (ratio) :2′ (ratio) |
| a 3 observed in traces (<5%) in the reaction mixture. b 4a isolated as a by-product (15%). c Methyl ketones isolated as by-products: 4b′ (R = n-hexyl, 16%), 4e′ (R = (MeO2C)2CHCH2, 17%), 4f′ (R = CH2-N-phthalamide, 32%). d Run without water (0.08 M). e Deprotection of one Boc- was observed under reaction conditions. f 100% conversion; products not isolated due to volatility issues. g Reactions carried out at 30 °C for 60 h. | ||
| 1 | R = Cy 1a | 59, 2a:2a′ (3.7:1)b |
| 2 | R = n-hexyl, 1b | 48, 2b:2b′ (1.9:1)c |
| 3 | R = n-octyl, 1c | 47, 2c:2c′ (1.8:1)d |
| 4 | R = Ph, 1d | 62, 2d |
| 5 | R = (MeO2C)2CHCH2, 1e | 43, 2e:2e′ (3.8:1)c |
| 6 | R = CH2-N-phthalamide, 1f | 48, 2f; 7, 2f′c |
| 7 | R = (Boc)2NCH2, 1g | 38, 2ge |
| 8 | R = R′ = Me, 1h |
2h:2h′ (3.7:1)f |
| 9 | R = Ph, R′′ = n-octyl, 1i | 76, 2i |
| 10 | R = Bn, R′′ = n-Pr, 1j | 67, 2j:2j′ (1.3:1) |
| 11 | R = Bn, R′′ = i-Pr, 1k | 55, 2k:2k′ (1.67:1) |
| 12 | R = Ph(CH2)2, R′′ = n-Pr, 1l | 70, 2l:2l′ (1:1.13) |
| 13 | R = Ph(CH2)2, R′′ = i-Pr, 1m | 80, 2m:2m′ (1:1) |
| 14g | R = p-CF3Ph, 1n | 74, 2n |
| 15g | R = p-CF3Ph, R′′ = n-Pr, 1o | 67, 2o |
| 16 | R = p-ClPh, R′′ = n-octyl, 1p | 72, 2p |
The reaction works for mono- and disubstituted allenes with different functionalities, but functional groups sensitive to acid are not well tolerated. Allyl azides 2 from the attack to the less substituted carbon of the allene were obtained as the major or only product in moderate to good yields. The regioselectivity of the reaction seems to be dominated by electronic effects: allenes directly substituted with a phenyl group gave only one regioisomer (entries 4, 9, 14, 15 and 16, Table 1). However, when the phenyl group was moved away from the allenic system (entries 10 to 13, Table 1), both regioisomers were observed, even in the case of bulkier substituents (R′′ = i-Pr, entries 11 and 13).
It is known that allyl azides equilibrate in solution, even at room temperature, by a [3,3] sigmatropic rearrangement.19 Monitoring the reaction of allene 1a by 1H NMR we observed that although both allyl azides are formed in the gold-catalysed reaction, the concentration of allyl azide 2a′ increases at the beginning and then decreases (2a′ = 31% at 1 h, 2a′ = 17%, at 3 h), isomerising into 2a during the reaction time.20 We postulate that amide 4a comes from the gold-catalysed Schmidt reaction of the methyl ketone,21 formed by protonation of the allene at the terminal position and attack of H2O, with the in situ formed HN3.
To investigate the possible formation of LAuN3 complexes,22 we carried out a preliminary NMR study: three stoichiometric experiments were performed at room temperature in which the order of addition of the components was varied. 1H and 31P NMR spectra were recorded after each addition (Scheme 3).
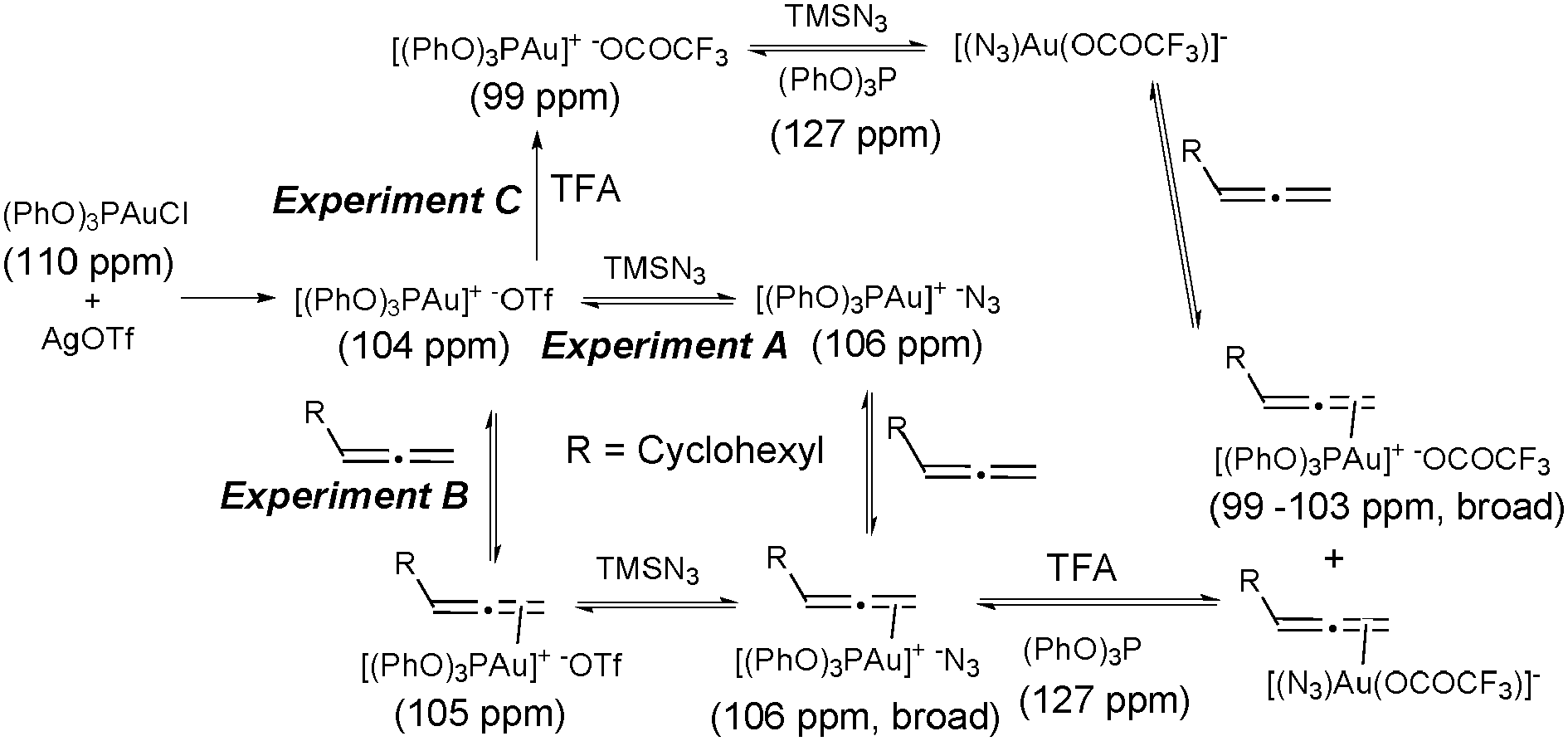 | ||
| Scheme 3 31P NMR-profile for stoichiometric experiments. | ||
Comparison of the reaction profile by analysis of the 31P NMR spectra in experiments A and B, where the addition of the allene and the azide were reversed, suggests that the azide coordinates to the gold in both cases, even when the allene is present in the reaction, and therefore an inner-sphere mechanism cannot be ruled out.23 Interestingly, after addition of TFA both reactions exhibited a broad signal in 31P NMR around 100 ppm together with a sharp singlet at 127 ppm from the free (PhO)3P, which grew over the reaction time.24 This suggests the CF3CO2− is capable of displacing either the (PhO)3P or the N3 from the gold to reach a complex equilibrium where at least three allene–gold complexes might be involved with different ligands/counterions.25 The exchange of TfO− by CF3CO2− and the displacement of (PhO)3P were also observed in experiment C, where TFA was added first. Further shift of the 31P NMR signal after addition of the TFA suggests the formation of the complex (PhO)3PAu+·−OCOCF3. The signal from free (PhO)3P was also observed in this reaction when azide was added, suggesting an equilibrium with the complex [(N3)Au(OCOCF3)]−. When the allene was added to this mixture a similar reaction profile to the experiments A and B was observed. Analysing the signals of the 1H NMR spectra we could not observe the proposed vinyl–gold intermediate, but clean conversion to the products, suggesting fast protonolysis. A similar rate of formation of the different products was observed in all experiments after all the components had been added, implying similar catalytic species involved after addition of the acid and suggesting that coordination with the allene and/or the N3− might be the rate limiting step for the reaction. When we followed the reaction using NMR under catalytic conditions, a broad signal around 100 ppm, and free (PhO)3P (127 ppm) were observed in 31P NMR spectra, which suggests that the complicated equilibrium between the complexes shown in Scheme 3 might be existing under catalytic conditions.26
The important utility of this transformation is illustrated by the straightforward synthesis of iodinated allyl azides. By using NIS as the electrophile in the gold-catalysed azidation of allenes,14,27 an extra functionality can be introduced in one step, making this method a very useful tool for the orthogonal functionalization of simple molecules into complex structures. The reaction of allene 1a with TMSN3, in the presence of NIS using different gold complexes afforded the vinyl iodide 5a as the only regioisomer (Scheme 4a). Vinyl iodide 5c was obtained in moderate yield using the NHC–Au catalyst (Scheme 4b).28
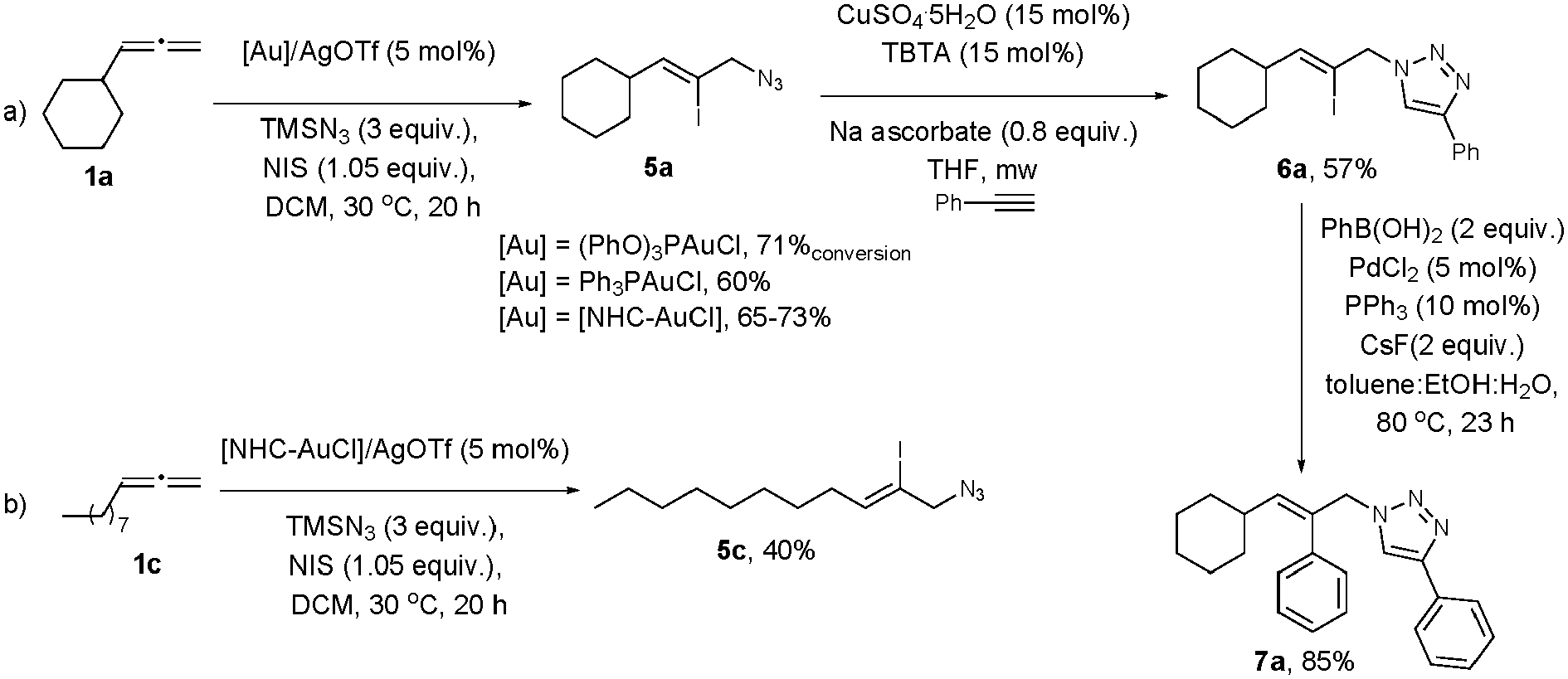 | ||
| Scheme 4 Orthogonal functionalization of allenes using the gold-catalysed azidation methodology. | ||
To further demonstrate the potential of this method as a highly modular approach, we carried out the synthesis of derivatives 6a and 7a by using a click29 and a Suzuki–Miyaura cross-coupling30 reaction with product 5a (Scheme 4a).
In conclusion, we have found that the hydroazidation of allenes to obtain allyl azides is possible by using (PhO)3PAuCl in the presence of TMSN3 and TFA. The reaction works well for substituted allenes, but functional groups sensitive to acid are not well tolerated. Preliminary mechanistic studies point out to a complex mechanism with equilibrium between several allene–gold–azide complexes, and an unusual displacement of the phosphite ligand from the gold-coordination sphere. Further investigation into this and a more detailed mechanistic study is underway in our laboratories. Further transformations involving the azide and diverse cross-couplings at different stages of the process are envisioned31 and will be of general interest to the synthetic chemistry community.
The authors would like to thank Prof. Hashmi for his support and the NHC–gold complexes. Funding by the University of East Anglia is gratefully acknowledged. This work was supported by the A-I Chem Channel program, selected by the European INTERREG IV A France (Channel) – England Cross-border cooperation Programme, co-financed by ERDF. The authors thank the EPSRC National Mass Spectrometry Service Centre, Swansea, for the accurate HRMS analysis.
Notes and references
- For selected examples, see: (a) A. Fürstner, M. Bindl and L. Jean, Angew. Chem., Int. Ed., 2007, 46, 9275–9278 CrossRef PubMed; (b) M. E. Kopach, M. M. Murray, T. M. Braden, M. E. Kobierski and O. L. Williams, Org. Process Res. Dev., 2009, 13, 152–160 CrossRef CAS.
- (a) S.-I. Murahashi, Y. Tanigawa, Y. Imada and Y. Taniguchi, Tetrahedron Lett., 1986, 27, 227–230 CrossRef CAS; (b) W. Zhou, J. Xu, L. Zhang and N. Jiao, Synlett, 2011, 887–890 CAS.
- For selected examples, see: (a) B. Sreedhar, P. S. Reddy and N. S. Kumar, Tetrahedron Lett., 2006, 47, 3055–3058 CrossRef CAS; (b) A. Mishra, S. Hutait, S. Bhowmik, N. Rastogi, R. Roy and S. Batra, Synthesis, 2010, 2731–2748 CAS.
- For selected examples, see: (a) K. Nishiyama and H. Karigomi, Chem. Lett., 1982, 1477–1478 CrossRef CAS; (b) S. G. Alvarez and M. T. Alvarez, Synthesis, 1997, 413–414 CrossRef CAS; (c) T. Kanai, Y. Kanagawa and Y. Ishii, J. Org. Chem., 1990, 55, 3274–3277 CrossRef CAS; (d) A. Jayanthi, V. K. Gumaste and A. R. A. S. Deshmukh, Synlett, 2004, 979–982 CAS.
- (a) W. Zhou, J. Xu, L. Zhang and N. Jiao, Synlett, 2011, 887–890 CAS , and references therein; (b) A. V. Malkov, P. Spoor, V. Vinader and P. Kocovsky, J. Org. Chem., 1999, 64, 5308–5311 CrossRef CAS.
- M. P. Muñoz, Org. Biomol. Chem., 2012, 10, 3584–3594 Search PubMed and references therein.
- For selected examples, see: (a) N. Nishina and Y. Yamamoto, Tetrahedron, 2009, 64, 1799–1808 CrossRef; (b) Z. J. Wang, D. Benitez, E. Tkatchouk, W. A. Goodward III and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 13064–13071 CrossRef CAS PubMed.
- For selected examples, see: (a) K. L. Toups, G. T. Liu and R. A. Widenhoefer, J. Organomet. Chem., 2009, 694, 571–575 CrossRef CAS PubMed; (b) M. A. Tarselli, A. Liu and M. R. Gagné, Tetrahedron, 2009, 65, 1785–1789 CrossRef CAS PubMed; (c) Z. Fang, C. Fu and S. Ma, Chem.–Eur. J., 2010, 16, 3910–3913 CrossRef CAS PubMed.
- For selected examples of the use of azides in gold-catalysis, see: (a) D. J. Gorin, N. R. David and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 11260–11261 CrossRef CAS PubMed; (b) Y. Sawama, Y. Sawama and N. Krause, Org. Lett., 2009, 11, 5034–5037 CrossRef CAS PubMed; (c) Z. Huo, I. D. Gridnev and Y. Yamamoto, J. Org. Chem., 2010, 75, 1266–1270 CrossRef CAS PubMed; (d) Z. Y. Yan, Y. Xiao and L. Zhang, Angew. Chem., Int. Ed., 2012, 51, 8624–8627 CrossRef CAS PubMed.
- For selected examples of reaction of allenes and nucleophilic azides, see: (a) H.-M. Chang and C.-H. Cheng, J. Chem. Soc., Perkin Trans. 1, 2000, 3799–3807 RSC; (b) H.-M. Chang, X. Huang, R. Shen and T. Zhang, J. Org. Chem., 2007, 72, 1534–1537 CrossRef PubMed.
- For selected examples of hydroazidation of alkenes, see: (a) I. Adamo, F. Benedetti, F. Berti and P. Campaner, Org. Lett., 2006, 8, 51–54 CrossRef CAS PubMed; (b) J. Waser, B. Gaspar, H. Nambu and E. M. Carreira, J. Am. Chem. Soc., 2006, 128, 11693–11712 CrossRef CAS PubMed.
- Ullmann's Encyclopedia of Industrial Chemistry, VCH Verlag, Weinheim, 1989, vol. A13, pp. 193–197 Search PubMed.
- For selected examples of in situ generation of HN3 using TMSN3, see: (a) G. W. Breton, K. A. Daus and P. J. Kropp, J. Org. Chem., 1992, 57, 6646–6649 CrossRef CAS; (b) H. Schäfer, W. Saak and M. Weidenbruch, J. Organomet. Chem., 2000, 604, 211–213 CrossRef.
- A. S. K. Hashmi, T. D. Ramamurthi, M. H. Todd, A. S.-K. Tsang and K. Graft, Aust. J. Chem., 2010, 63, 1619–1626 CrossRef CAS , and references therein.
- All the control experiments gave negative results (see ESI† for details).
- For silver effect in gold catalysis, see: D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedow, H. Zhang, X. Liu, J. L. Pettersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012–9019 CrossRef CAS PubMed.
- Gold-catalysed reaction of allenes in the presence of water gives allyl alcohols: Z. Zang, S. D. Lee, A. S. Fisher and R. A. Widenhoefer, Tetrahedron, 2009, 65, 1794–1798 CrossRef PubMed . However, despite the high excess of water, we do not observe significant amounts of hydration of the allene in our reaction, and formation of 3 is minimised.
- Different proton sources and additive to modulate the pH were tested, being TFA (3 eq.) plus water (5 eq.) the best combination for yield and selectivity. See Tables S2 and S3, in ESI†.
- (a) A. Gagneux, S. Winstein and W. G. Young, J. Am. Chem. Soc., 1960, 82, 5956–5957 CrossRef CAS; (b) A. K. Feldman, B. Colasson, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 13444–13445 CrossRef CAS PubMed.
- This seems to be in agreement with reported examples using excess of alcohol as nucleophile, where the kinetic product is the attack to the more substituted carbon, and a gold-catalysed isomerisation then occurs to give the thermodynamic less substituted allyl ethers. (a) M. S. Hadfield and A.-L. Lee, Org. Lett., 2010, 12, 484–487 CrossRef CAS PubMed; (b) R. S. Paton and F. Maseras, Org. Lett., 2009, 11, 2237–2240 CrossRef CAS PubMed.
- K. F. Z. Schmidt, Angew. Chem., Int. Ed. Engl., 1923, 36, 511 Search PubMed.
- D. V. Partyka, T. J. Robilotto, M. Zeller, A. D. Hunter and T. G. Gray, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14293–14297 CrossRef CAS PubMed , and references therein.
- Independently synthesised (PhO)3PAuN3 has a 31P NMR signal at 105.80 ppm, and IR sharp band at 2060 cm−1 (νas N3−). This complex resulted to be very unstable and could not be tested as catalyst in the reaction.
- Broad signals in 31P NMR might also indicate di-aurated species: T. J. Brown, D. Weber, M. R. Gagné and R. A. Widenhoefer, J. Am. Chem. Soc., 2012, 134, 9134–9137 CrossRef CAS PubMed.
- Phosphite ligands bind more weakly to Au than phosphines, see: G. C. Fortman and S. P. Nolan, Organometallics, 2010, 29, 4579–4583 CrossRef CAS.
- When the reaction using 15 mol% of catalyst was followed by IR, only a signal corresponding to the product 2a:2a′ was observed in the azide region (ν = 2097 cm−1). If existent, the signal corresponding to the [Au–N3] complex would appear at 2060 cm−1, mostly overlapped with the allyl azide 2, and would be difficult to observed. Therefore, the possibility of involvement of this catalytic specie can not be totally ruled out at present (see ESI† for details).
- For a selected example of trapping vinyl–gold intermediates with NIS in allene chemistry, see: A. Heuer-Jungemann, R. G. McLaren, M. S. Hadfield and A.-L. Lee, Tetrahedron, 2011, 1609–1616 CrossRef CAS.
- A. Hassner and J. Keogh, J. Org. Chem., 1986, 51, 2767–2770 CrossRef CAS . When our reaction was carried out in the absence of gold or silver complexes, the starting material was recover unreacted, implying that no IN3 is formed in our case.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS , and references therein.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- Attempts to engage the viny–gold intermediate in oxidative cross-coupling have been so far unsuccessful; see: G. Zhang, Y. Peng, L. Cui and L. Zhang, Angew. Chem., Int. Ed., 2009, 48, 3112–3115 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc48017h |
| This journal is © The Royal Society of Chemistry 2014 |