 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Acyl hydrazides as acyl donors for the synthesis of diaryl and aryl alkyl ketones†
Ahmed R.
Akhbar
a,
Vijay
Chudasama
*a,
Richard J.
Fitzmaurice
a,
Lyn
Powell
b and
Stephen
Caddick
*a
aDepartment of Chemistry, University College London, London, WC1H 0AJ, UK. E-mail: VPEnterprise@ucl.ac.uk; v.chudasama@ucl.ac.uk; Tel: +44 (0)20 3108 5071
bAstraZeneca, Charter Way, Silk Road Business Park, Macclesfield, Cheshire SK10 2NA, UK
First published on 22nd November 2013
Abstract
In this communication we describe a novel strategy for the formation of valuable diaryl and aryl alkyl ketones from acyl hydrazides. A wide variety of ketones are prepared and the mild reaction conditions allow for the use of a range of functionalities, especially in the synthesis of diaryl ketones.
Diaryl and aryl alkyl ketones are important intermediates in the synthesis of various pharmaceuticals, natural products, agrochemicals and other functional materials.1 They are synthesised by numerous protocols, perhaps the most common of which is Friedel–Crafts acylation.1 Although this method has been useful for the synthesis of many ketones, it suffers from poor functional group tolerance, has untunable regioselectivity and requires the use of a stoichiometric amount of Lewis acid.2 Another common method to access ketones is the oxidation of secondary alcohols. However, this protocol often requires the use of a stoichiometric amount of oxidant and is commonly limited by poor functional group tolerance.3 The reaction of carboxylic acid derivatives such as anhydrides and acid chlorides with organometallic reagents tend to suffer from over-addition (resulting in the formation of tertiary alcohol products) and poor chemoselectivity.4 However, a notable exception in this class of derivatives are Weinreb amides.5 Treatment of a Weinreb amide with a suitable organometallic reagent provides ketones in high yields without significant over-addition. This selectivity is as a result of the formation of a stable, metal-chelate tetrahedral intermediate under the reaction conditions, which prevents over addition of nucleophile (Fig. 1).5 The tetrahedral intermediate is subsequently destroyed upon work-up to afford ketone.5
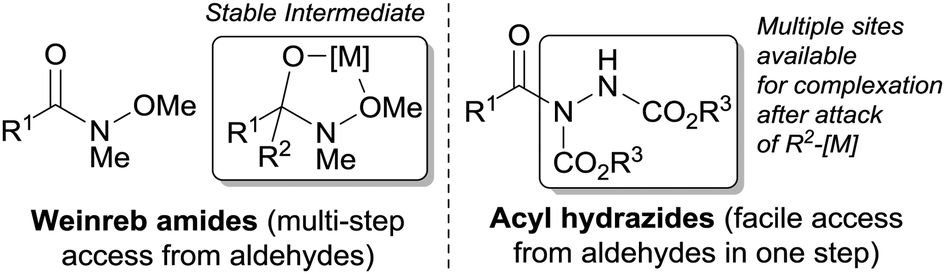 | ||
| Fig. 1 Comparison between Weinreb amides and acyl hydrazides. | ||
We have previously described simple and efficient methods for the hydroacylation of various radical acceptors using atmospheric oxygen as radical initiator.6 Most recently, we reported a highly efficient method for the functionalisation of aldehydes to acyl hydrazides via the hydroacylation of azo-dicarboxylates.6e In that report, we highlighted that acyl hydrazides may be employed as acyl donors for the synthesis of amides.6e Given the similarities between acyl hydrazides and Weinreb amides, the fact they have plenty of sites available for forming a stable metal chelate and have been shown to be highly stable and effective acyl donors, we were intrigued as to whether acyl hydrazides could serve as effective acyl donors for the synthesis of ketones. Such a transformation would be highly desirable due to the facile and mild way in which aldehydes can be readily transformed into acyl hydrazides in a single step.6e The conversion of an aldehyde to a Weinreb amide, in contrast, typically requires multiple steps and relatively harsh reaction conditions.5b
Investigations to explore the feasibility of converting acyl hydrazides into ketones were initiated by reaction of acyl hydrazide 1a with n-PnMgBr 2a (Table 1). Initially, we applied typical Weinreb ketone synthesis reaction conditions (−10 °C, THF, 1.2 eq. of RMgX) to affect formation of ketone 3aa.5b Encouragingly, reaction under these conditions afforded ketone 3aa, albeit in low yield, 21% (Table 1, entry 1). Also isolated from the reaction mixture were secondary and tertiary alcohols, 4 and 5, in 14% and 28% yield, respectively (Table 1, entry 1). Formation of secondary alcohol 4 may be rationalised by β-hydride elimination of a Grignard reagent complexed to ketone 3aa.7 Tertiary alcohol 5 is likely to have formed by addition of n-PnMgBr 2a to ketone 3aa formed in the reaction conditions.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Temp. (°C) | Time (h) | 2a (eq.) | Conversion 1aa (%) | 3aa (%) | 4 (%) | 5 (%) |
| a Determined by integration of 19F NMR relative to trifluorotoluene as an internal standard. | |||||||
| 1 | −10 | 0.5 | 1.2 | 84 | 21 | 14 | 28 |
| 2 | −10 | 0.5 | 2 | 100 | 0 | 12 | 42 |
| 3 | −40 | 0.5 | 1.2 | 64 | 41 | 10 | 10 |
| 4 | −40 | 0.5 | 2 | 95 | 32 | 11 | 21 |
| 5 | −78 | 0.5 | 1.2 | 34 | 21 | 2 | 0 |
| 6 | −78 | 0.5 | 2 | 74 | 64 | 4 | 2 |
| 7 | −78 | 1 | 2 | 77 | 61 | 5 | 1 |
| 8 | −78 | 2 | 2 | 75 | 60 | 3 | 2 |
| 9 | −78 | 3 | 2 | 80 | 62 | 4 | 2 |
| 10 | −78 | 0.5 | 2.5 | 89 | 78 | 7 | 3 |
| 11 | −78 | 0.5 | 3 | 91 | 72 | 7 | 6 |
| 12 | −78 | 0.5 | 4 | 92 | 71 | 8 | 6 |
| 13 | −78 | 0.5 | 5 | 94 | 69 | 12 | 5 |
| 14 | −78 | 0.5 | 6 | 100 | 72 | 11 | 8 |
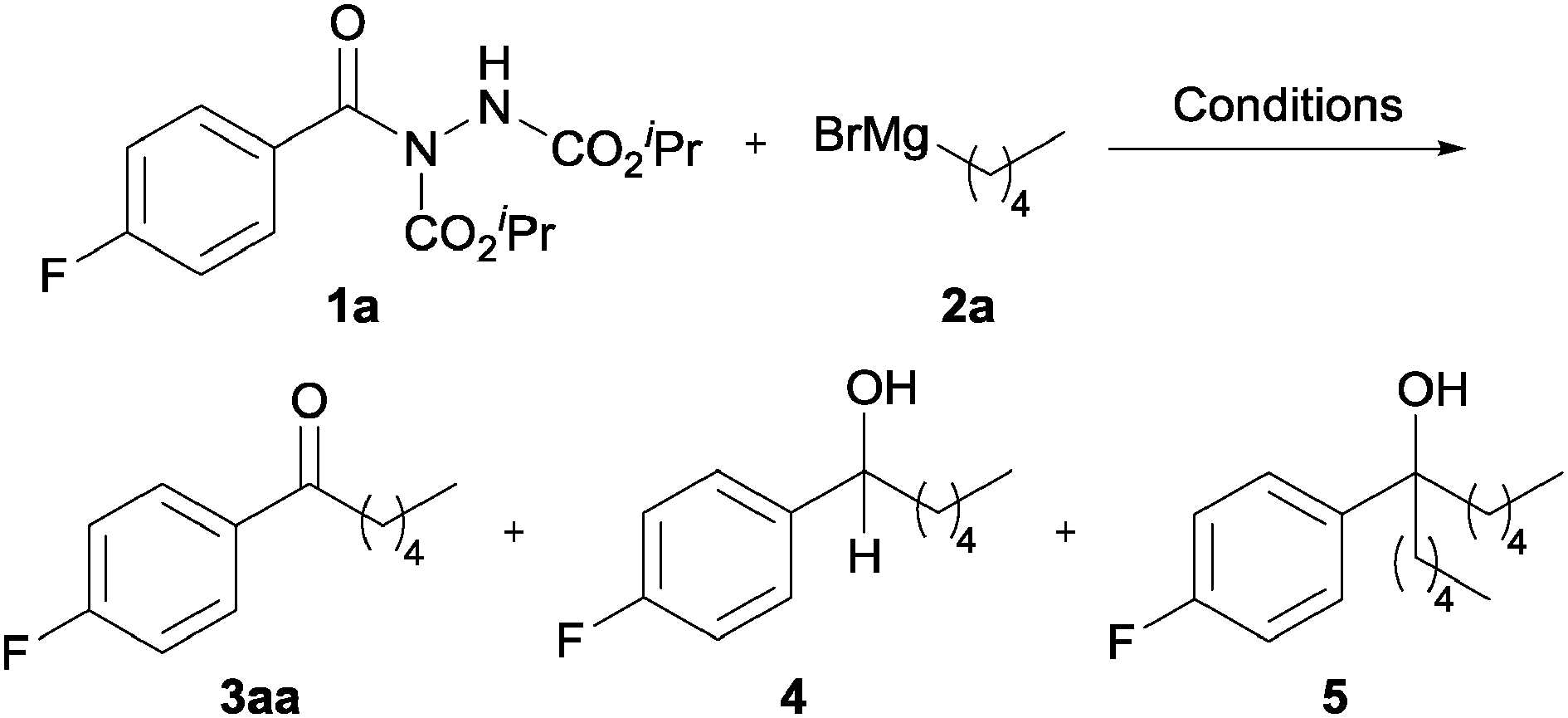
Perhaps as expected, the addition of greater equivalents of 2a led to an increase in the amount of tertiary alcohol, with no ketone being observed at all (Table 1, entry 2). We rationalised that reaction at a lower temperature would give better opportunity for a complex formed by addition of a Grignard onto an acyl hydrazide to be persistent. To investigate this, reaction of 1a with 1.2 and 2 equivalents of 2a was carried at −40 °C and −78 °C (Table 1, entries 3–6). Most pleasingly, decreasing the reaction temperature resulted in a decrease in the amount of 4 and 5, and a gradual increase in yield of ketone with maximum yield, 64%, being observed with 2 equivalents of 2a at −78 °C. In an attempt to further increase conversion and yield of ketone 3aa, the effect of increasing reaction time was explored (Table 1, entries 7–9). However, this had little effect on conversion and yield, with comparable yields being observed for reaction at −78 °C for 0.5 to 3 h. In a further attempt to increase yield of ketone, the use of a greater equivalents of 2a was explored (Table 1, entries 10–14). Gratifyingly, this afforded increasingly higher conversion of acyl hydrazide 1a (i.e. from 80% to 100%) with an excellent yield of ketone, 78%, being observed with 2.5 equivalents of 2a (Table 1, entry 10). Although using greater than 2.5 equivalents gave higher conversion, there was no increase in the yield of ketone (Table 1, entries 11–14).
We next applied our optimised conditions to the reaction of acyl hydrazide 1a with phenyl magnesium bromide 2b to evaluate the applicability of the conditions for the formation of diaryl ketones. Unfortunately, this only afforded a low yield of ketone 3ab, 7%, due to low conversion of 1a, 12%, perhaps as 2b is a less potent nucleophile than 2a. To combat this, addition of 2b at −78 °C was followed by warming to 0 °C. Pleasingly, this afforded ketone 3ab in 78% yield (Table 2, entry 1).
|
|
|||
|---|---|---|---|
| Entry | Acyl hydrazide 1, R = | Isolated yield 3/%, R′ = | |
|
|
|
||
| Conditions:a −78 °C, THF, 30 min.b −78 °C to 0 °C, THF, 1 h. | |||
| 1 |
|
78, 3aa | 78, 3ab |
| 2 |
|
77, 3ba | 87, 3bb |
| 3 |
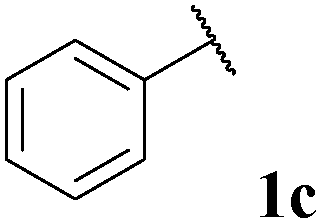
|
71, 3ca | 73, 3cb |
| 4 |
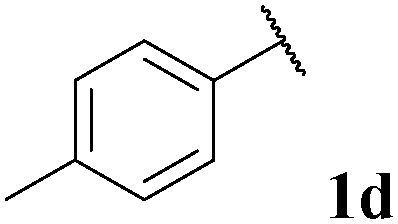
|
68, 3da | 70, 3db |
| 5 |
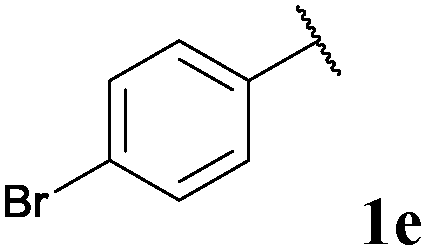
|
79, 3ea | 71, 3eb |
| 6 |
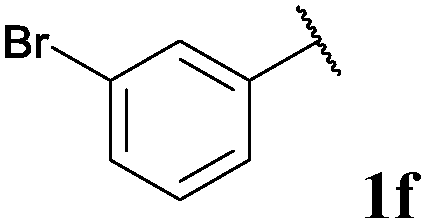
|
72, 3fa | 92, 3fb |
| 7 |
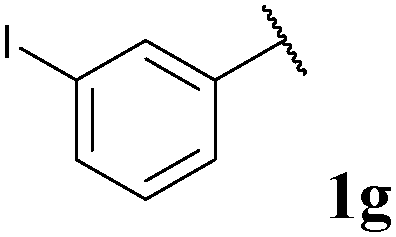
|
76, 3ga | 72, 3gb |
| 8 |
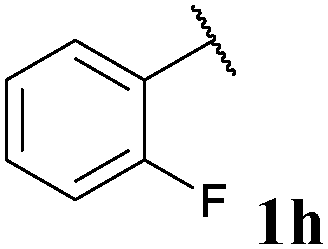
|
36, 3ha | 74, 3hb |
| 9 |
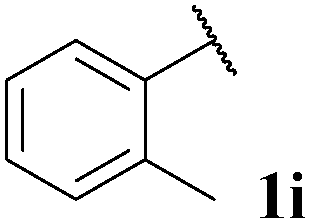
|
44, 3ia | 73, 3ib |
| 10 |
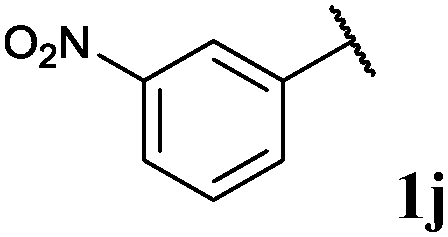
|
0, 3ja | 77, 3jb |
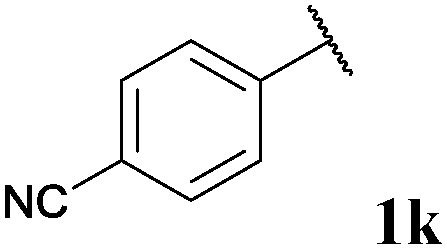
| 11 |
|
0, 3ka | 76, 3kb |
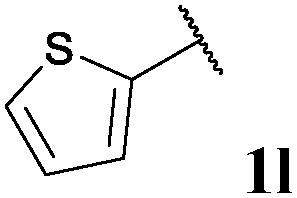
| 12 |
|
74, 3la | 76, 3lb |
With optimised conditions in-hand we took the opportunity to investigate the applicability of our protocol for the formation of ketones from various aryl acyl hydrazides. All starting hydrazides (1a–l) were prepared using our previously reported facile method for the hydroacylation of DIAD in good yields.6e Each acyl hydrazide was then reacted with n-pentyl magnesium bromide 2a and phenyl magnesium bromide 2b (Table 2) under the optimised conditions developed for the formation of aryl alkyl and diaryl ketones, respectively. Encouragingly, good to excellent yields were observed for reaction of 2a and 2b with electron poor (Table 2, entries 1 and 2), electron neutral (Table 2, entry 3), and electron rich (Table 2, entry 4) aryl acyl hydrazides. It was also pleasing that bromo- and iodo-functionalised aryl acyl hydrazides were tolerant of the reaction conditions and afforded ketones in good yields (Table 2, entries 5–7). Although ortho-substitution was not so well tolerated on application of 2a, good yields were observed for the synthesis of diaryl ketones (Table 2, entries 8 and 9). Owing to the mildness of the reaction conditions required for effective conversion, we appraised the use of acyl hydrazides bearing functionalities that were not tolerant of Weinreb amide ketone synthesis conditions, i.e. nitro- and cyano-groups (Table 2, entries 10 and 11). Although no product was isolated on reaction with n-PnMgBr 2a, diaryl ketones 3jb and 3kb were isolated in excellent yields on application of PhMgBr 2b, demonstrating to some extent the mildness of our protocol. We also show that a heterocyclic acyl hydrazide, 1l, gave good yields of aryl alkyl and diaryl ketone upon reaction with n-PnMgBr 2a and PhMgBr 2b, respectively (Table 2, entry 12).
Having demonstrated good tolerance of acyl hydrazides for the formation of diaryl and aryl alkyl ketones on application of Grignards 2a and 2b, we explored the tolerance of other Grignard reagents (Fig. 2). To do this, acyl hydrazide 1a was reacted with alkynyl, thiophenyl and isopropyl magnesium bromides. Gratifyingly, this afforded excellent yields of ketones 6–8, thus demonstrating the flexibility of our protocol with respect to the nature of the Grignard reagent.
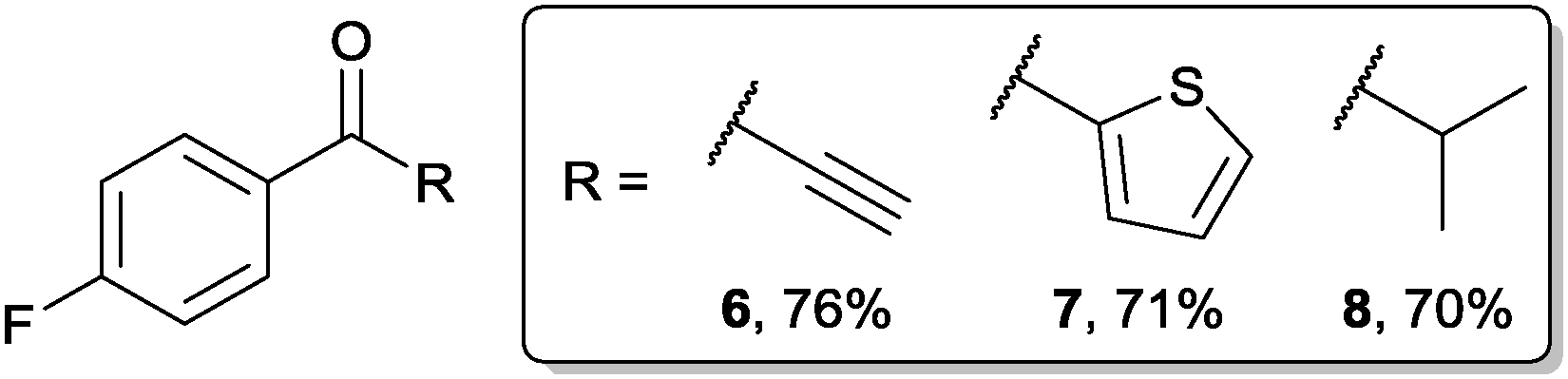 | ||
| Fig. 2 Formation of various ketones using our protocol. | ||
In order for the yield of ketone to be so high we rationalised that there must be some complexation to stabilise the adduct formed upon nucleophilic addition to acyl hydrazide. We rationalised that complexation could be affected through one of three possibilities (Fig. 3): (i) deprotonated hydrazide, 9; (ii) ester carbonyl, 10; or (iii) N-lone pair, 11.
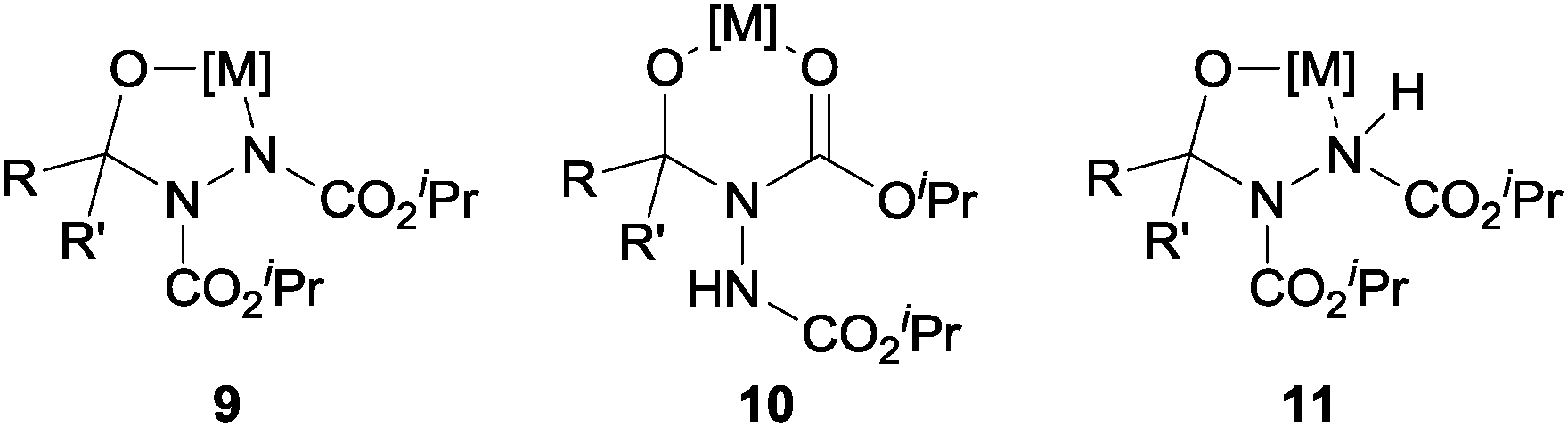 | ||
| Fig. 3 Potential stable intermediates. | ||
To appraise the feasibility of each of these postulated intermediates we initially explored whether an acyl hydrazide would be deprotonated under the reaction conditions owing to the acidity of the N–H bond. To do this, acyl hydrazide 1a was reacted with a single equivalent of Grignard 2a, followed by addition of methyl iodide, all at −78 °C, and then quenching with ammonium chloride (Scheme 1a). As this resulted in the formation of a significant amount of methylated hydrazide 12, 87%, we felt it appropriate to conclude that an acyl hydrazide would be significantly deprotonated under the reaction conditions, thus disfavouring the likelihood of intermediate 11. Further support for the first equivalent of Grignard acting as base was obtained by reaction of acyl hydrazide 1a with n-PnMgBr 2a (1 eq.) followed by addition of PhMgBr 2b (1.5 eq.). This afforded primarily diaryl ketone 3ab, 71%, and a modest amount of 3aa.
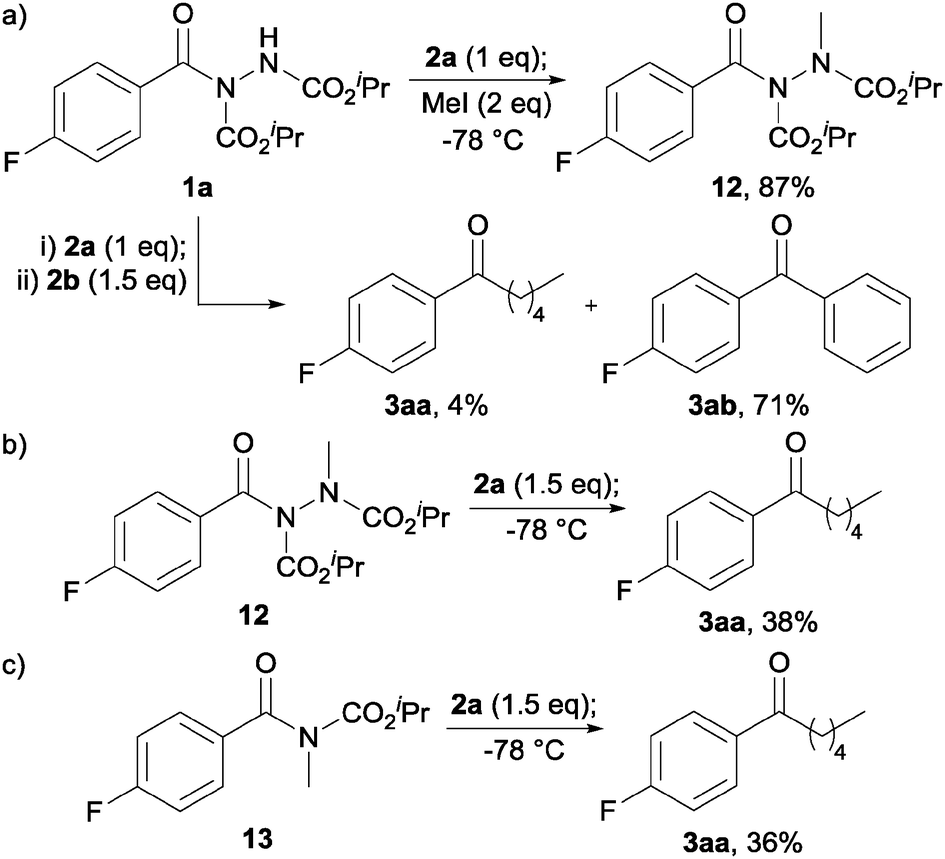 | ||
| Scheme 1 Reaction of: (a) hydrazide 1a with 2a then MeI, and with 2a followed by 2b; (b) hydrazide 12 with 2a; and (c) carbamate 13 with 2a. | ||
We next evaluated the importance of deprotonation for efficient ketone synthesis to transpire by reaction of methylated hydrazide 12 with 1.5 equivalents of 2a (Scheme 1b). This resulted in a poor yield of ketone, 38%, with a significant amount of tertiary alcohol being observed (20%), perhaps indicating that deprotonation is important for efficient ketone formation to ensue. Finally, we synthesised carbamate 13 and reacted it with 1.5 equivalents of 2a to determine if carbonyl complexation may play a key role in forming a stable intermediate, especially in view of a report on the reaction of Grignard reagents with N-Boc protected β-, γ- and δ-lactams for the synthesis of ketones.8 However, reaction of 13 with 2a only afforded ketone in 36% yield, again due to the formation of a significant amount of tertiary alcohol (Scheme 1c). Thus, it seems reasonable to conclude the high yield of ketone observed in our protocol is most likely due to the formation of a persistent intermediate of the form of 9 (see Fig. 3).
In summary, the work described herein represents, to the best of our knowledge, the first examples of the use of acyl hydrazides as acyl donors for the synthesis of ketones. This is particularly exciting in view of the facile manner in which acyl hydrazides may be prepared from various aldehydes6e,9 or otherwise.10 Moreover, ketone formation has been shown to proceed under mild conditions, which has allowed for the tolerance of sensitive functional groups such as nitrile and nitro for the synthesis of diaryl ketones, and various Grignard reagents. We will now seek to evaluate the applicability of our protocol for the formation of dialkyl ketones, and especially those derived from enantiopure alkyl acyl hydrazides.
Notes and references
- (a) S. Rahimipour, C. Palivan, F. Barbosa, I. Bilkis, Y. Koch, L. Weiner, M. Fridkin, Y. Mazur and G. J. Gescheidt, J. Am. Chem. Soc., 2003, 125, 1376 CrossRef CAS PubMed; (b) Y. Deng, Y.-W. Chin, H. Chai, W. J. Keller and A. D. Kinghorn, J. Nat. Prod., 2007, 70, 2049 CrossRef CAS PubMed; (c) M. Van de Putte, T. Roskams, J. R. Vandenheede, P. Agostinis and P. A. M. deWitte, Br. J. Cancer, 2005, 92, 1406 CrossRef CAS PubMed; (d) Y. Nishizuka, Nature, 1988, 334, 661 CrossRef CAS PubMed; (e) X.-Q. Ding, E. Lindstrom and R. Hakanson, Pharmacol. Toxicol., 1997, 81, 232 CrossRef CAS; (f) A. M. Prince, D. Pascual, D. Meruelo, L. Liebes, Y. Mazur, E. Dubovi, M. Mandel and G. Lavie, Photochem. Photobiol., 2000, 71, 188 CrossRef CAS; (g) X. Jia, S. Zhang, W. Wang, F. Luo and J. Cheng, Org. Lett., 2009, 11, 3120 CrossRef CAS PubMed.
- G. A. Olah, Friedel–Crafts Chemistry, Wiley, New York, 1973 Search PubMed.
- G. Tojo and M. Fernandez, Oxidation of Alcohols to Aldehyde and Ketones: A Guide to Current Common Practice, Springer, 2006 Search PubMed.
- J. March, Advanced Organic Chemistry, Wiley, New York, 3rd edn, 1985, pp. 433–435 and 824–827 CrossRef CAS; R. K. Dieter, Tetrahedron, 1999, 55, 4177 CrossRef CAS.
- (a) S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815 CrossRef CAS; (b) S. Balasubramaniam and I. S. Aidhen, Synthesis, 2008, 3707 CAS.
- (a) R. J. Fitzmaurice, J. M. Ahern and S. Caddick, Org. Biomol. Chem., 2009, 7, 235 RSC; (b) V. Chudasama, R. J. Fitzmaurice, J. M. Ahern and S. Caddick, Chem. Commun., 2010, 46, 133 RSC; (c) V. Chudasama, R. J. Fitzmaurice and S. Caddick, Nat. Chem., 2010, 2, 592 CrossRef CAS PubMed; (d) V. Chudasama, J. M. Ahern, R. J. Fitzmaurice and S. Caddick, Tetrahedron Lett., 2011, 52, 1067 CrossRef CAS PubMed; (e) V. Chudasama, R. J. Fitzmaurice, D. V. Dhokia, J. M. Ahern and S. Caddick, Chem. Commun., 2011, 47, 3269 RSC; (f) V. Chudasama, A. R. Akhbar, K. A. Bahou, R. J. Fitzmaurice and S. Caddick, Org. Biomol. Chem., 2013, 11, 7301 RSC.
- H. Maeda, J. Okamoto and H. Ohmori, Tetrahedron Lett., 1996, 37, 5381 CrossRef CAS.
- A. Giovannini, D. Savoia and A. U. Ronchi, J. Org. Chem., 1989, 54, 228 CrossRef CAS.
- Q. Zhang, E. Parker, A. D. Headley and B. Ni, Synlett, 2010, 2453 Search PubMed; B. Ni, Q. Zhang, S. Garre and A. D. Headley, Adv. Synth. Catal., 2009, 351, 875 CrossRef CAS.
- I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, S. Montanaro and M. Fagnoni, Org. Lett., 2013, 15, 2554 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all novel compounds and compounds prepared by a novel protocols. See DOI: 10.1039/c3cc47967f |
| This journal is © The Royal Society of Chemistry 2014 |