 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New catalytic model systems of tyrosinase: fine tuning of the reactivity with pyrazole-based N-donor ligands†
Jessica Nadine
Hamann
and
Felix
Tuczek
*
Christian-Albrechts-Universität zu Kiel, Max-Eyth-Straße 2, 24118 Kiel, Germany. E-mail: ftuczek@ac.uni-kiel.de; Fax: +49-(0)431-1520; Tel: +49-(0)431-1410
First published on 9th January 2014
Abstract
Two new Cu(I) complexes have been synthesized and investigated as model systems of the enzyme tyrosinase. The corresponding ligands are based on a combination of an imine function with two different pyrazole groups. The reactivity of the prepared systems with respect to the conversion of monophenols to the corresponding ortho-quinones is investigated. The resulting data are compared to results obtained for other catalytic model systems of tyrosinase.
The ubiquitous type 3 copper enzyme tyrosinase (Ty) catalyses the formation of ortho-quinones, starting from monophenols. The conversion of tyrosine to dopaquinone occurs in a two-step process, starting with an aromatic hydroxylation which is followed by two-electron oxidation.1,2 The formed dopaquinone polymerises spontaneously to the important pigment melanin.3,4 The active site of tyrosinase contains a binuclear copper centre, wherein each copper ion is coordinated by three histidine residues.5,6 In 2006 Matoba and coworkers published the first crystal structure of a tyrosinase derived from the bacterium Streptomyces castaneoglobisporus.5 In the meantime more tyrosinases have been structurally characterized.7
Hemocyanins (Hc) and catechol oxidases (CO) are also important type 3 copper proteins, but exhibit reactivities different to tyrosinase. Hemocyanins mediate the oxygen transport in arthropods and molluscs whereas catechol oxidases are responsible for the oxidation of catechols to the corresponding ortho-quinones.6,8,9 All of these copper type 3 enzymes have very similar active sites and bind dioxygen as peroxide in a typical μ2–η2:η2 (side-on bridging) geometry. In the oxy form the oxidation state of copper ions changes from +I to +II.1
Tyrosinase exhibits monophenolase and diphenolase activity. The monophenolase cycle involves hydroxylation of monophenolic substrates to catechols, which are released as ortho-quinones (Scheme 1).1,6,10 In recent years, many low-molecular copper complexes have been synthesized as model systems of tyrosinase.1,11 These compounds can be divided into systems which hydroxylate the ligand framework12–16 and those which are able to convert external monophenols to the corresponding ortho-quinones.11,17 The first model system exhibiting the latter reactivity in a catalytic fashion was published by Réglier and coworkers.18 This binuclear copper(I) complex generates 3,5-di-tert-butyl-o-quinone (DTBQ) from 2,4-di-tert-butyl phenol (DTBP-H) with a turnover number (TON) of 16. The formation of ortho-quinone was detected by using UV/Vis spectroscopy, because ortho-quinones show an intense absorption band in the range of 400–420 nm.19 In 1991 a further system exhibiting stoichiometric as well as weakly catalytic monooxygenation of external phenols was reported by Casella and coworkers.17 This year Herres-Pawlis et al. presented a new copper(I) catalyst mediating the hydroxylation of monophenols via a well-characterized peroxo intermediate.20
 | ||
| Scheme 1 Conversion of monophenolic substrates to ortho-quinones via catechols (monophenolase and diphenolase activity, respectively). | ||
In 2010 our group reported the first catalytic model system based on a mononuclear copper(I) complex.11 This [Cu(I)Lpy1(CH3CN)2]PF6 system (cf.Scheme 2) was found to generate DTBQ from DTBP-H with a TON of 18 after 8 hours. More recently we presented a second model system containing a benzimidazole moiety.21 With the new Lbzm1 system, the TON could be increased to 31, along with a significantly increased turnover frequency.21 To further investigate the influence of the heterocyclic group on the reactive properties of our system, we developed two new pyrazole based copper(I) complexes as model systems.
 | ||
| Scheme 2 (a) Ligand Lpy1 of the first mononuclear catalytic model system, (b) ligand Lbzm1 and ligands Lhpz1 and Lhpz2 of this study (c and d).11,21 | ||
The ligands of both systems contain an imine function terminated by a tert-butyl residue; they only differ with respect to the substituents of the pyrazole moiety (Lhpz1 and Lhpz2; Scheme 2).
Synthesis and reactivity of the new Cu(I) complexes. The two ligands Lhpz1 and Lhpz2 were prepared in several steps by using similar procedures (Scheme 3 and ESI†). For the aminoethylation of the pyrazoles a modification of a literature procedure was used.22 The corresponding copper(I) complexes were prepared from tetrakis(acetonitrile)copper(I) hexafluorophosphate under anaerobic conditions.
 | ||
| Scheme 3 Syntheses and metalation of the new ligands. | ||
To investigate the catalytic activity of the pyrazole-based model systems in situ UV/Vis spectroscopy was applied. To this end a 500 μM solution of the respective copper(I) complex in dichloromethane was prepared and 50 eq. of DTBP-H and 100 eq. of triethylamine were added. Subsequent oxygenation at ambient temperature was found to result in the formation of DTBQ, as indicated by an absorption band appearing at 407 nm (Fig. 1).
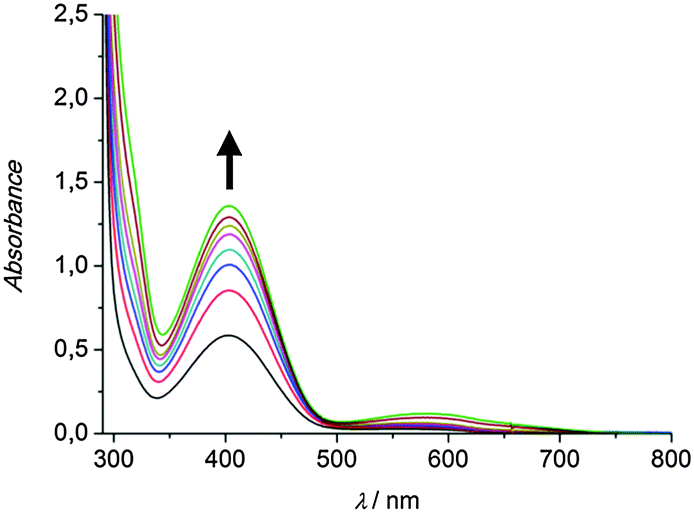 | ||
| Fig. 1 UV/Vis spectra of a 500 μM solution of the complex [Cu(I)Lhpz1(CH3CN)2]PF6 in CH2Cl2 after addition of 50 eq. DTBP-H, 100 eq. NEt3 and oxygenation between 15 min and 6 h; l = 1 mm. | ||
As evident from Fig. 1, the complex [Cu(I)Lhpz1(CH3CN)2]PF6 catalyses the formation of 3,5-di-tert-butyl-o-quinone (DTBQ) from the corresponding phenol DTBP-H. For the determination of the turnover number (TON), an extinction coefficient of ε = 1830 M−1 cm−1 at 407 nm was applied.19 Oxygenation of DTBP-H occurred very fast during the first 100 minutes of reaction and then slowed down, leading to a TON of 29 after 6 hours.
The oxygenation was also performed using the complex [Cu(I)Lhpz2(CH3CN)2]PF6. In analogy to the Lhpz1 system, the formation of DTBQ happened quickly during the first two hours, reaching a TON of 23 after 6 hours. In Fig. 2 the TON and TOF (turnover frequency) of both systems are compared. Importantly, the Lhpz1 system exhibits both a higher TON (29) and a higher TOF (0.85 min−1 after 15 min) as compared to its Lhpz2 counterpart (TON = 23, TOF = 0.62 min−1). Both systems form most of the quinone during the first 120 min and become inactive after ∼6 hours.
 | ||
| Fig. 2 Turnover frequency as a function of time (15 min < t < 6 hours) for the systems Lhpz1 and Lhpz2; inset: their turnover number per dicopper unit as a function of time. | ||
In order to further prove the formation of DTBQ during the reaction of DTBP with molecular oxygen NMR spectra of the reaction mixture were recorded (Fig. 3). To this end the solutions were quenched with 6 M hydrochloric acid after 30 minutes of oxygenation and extracted with dichloromethane to eliminate the copper ions.
 | ||
| Fig. 3 1H-NMR spectrum of the oxygenation solution after quenching with HCl (organic phase) for the Lhpz1 system. Inset: resonances in the aliphatic region. | ||
In agreement with similar model systems the resulting NMR spectra revealed signals for DTBQ, DTBP-H and the C–C coupling product 3,3′,5,5′-tetra-tert-butyl-2,2′-biphenol (“biphenol”, Fig. 3).11,21 The signals of DTBP-H, DTBQ and the biphenol are found at a ratio of 51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18:31. This result agrees with the TON of 18 measured after 30 minutes of oxygenation (Fig. 2). For the Lhpz2 system the corresponding ratio was obtained as 70:15:15 (cf. ESI†).
:18:31. This result agrees with the TON of 18 measured after 30 minutes of oxygenation (Fig. 2). For the Lhpz2 system the corresponding ratio was obtained as 70:15:15 (cf. ESI†).
Related small-molecule model systems of tyrosinase investigated before (cf.Scheme 2) have already indicated an important influence of the heterocyclic N-donor group on the catalytic activity.11,21 Specifically, the CuLbzm1 complex was found to mediate the conversion of DTBP-H to DTBQ with a TON of 31, 72% higher than that of CuLpy1, and a TOF which was about twice as large as that of the latter complex. This was in part attributed to the well-documented capability of copper–benzimidazole complexes to mediate the ortho-hydroxylation of phenolic substrates.17 The new pyrazole-based systems range between the original Lpy1 system and the Lbzm1 system; i.e., the methylated Lhpz2 catalyst is somewhat more active than Lpy1 and the unsubstituted Lhpz1 catalyst is almost as active as Lbzm1. To explain this trend we suggest that electron-rich N-donor ligands promote the hydroxylation reaction by making the peroxo intermediate less stable (Scheme 4, compound 3); i.e. the least reactive pyridine-based system would form the most stable peroxo intermediate whereas the stability of the peroxo intermediate is decreased for the more reactive benzimidazole- or pyrazole based catalysts. In fact, we succeeded in observing the peroxo adduct for the Lpy1 complex at low temperatures, in contrast to the latter systems (cf. ESI†). A second possible influence on the reaction rate emerges from the two-electron oxidation of the catecholate adduct 4, leading to the quinone.
 | ||
| Scheme 4 Proposed catalytic cycle for investigated systems Lhpz1 and Lhpz2 in analogy to model systems Lpy1 and Lbzm1.11,21 | ||
With respect to the system without substituted pyrazole (Lhpz1; TON = 29), the TON decreases to 23 for the Lhpz2 system. In agreement with the above considerations we attribute this effect to (i) increased stabilization of the peroxo adduct and (ii) an increased steric hindrance with respect to coordination of the substrate (phenol) to the peroxo intermediate (Scheme 4).
Two new mononuclear copper(I) complexes containing ligands with pyrazole groups were synthesized as model systems of the enzyme tyrosinase and investigated regarding the conversion of the external substrate DTBP-H to DTBQ, using UV/Vis and NMR spectroscopy. Importantly, the Lhpz1 system (TON = 29) was found to be more reactive than its Lhpz2 counterpart (TON = 23; Table 1).
| System | Lpy1 | Lhpz2 | Lhpz1 | Lbzm1 |
|---|---|---|---|---|
| TON | 22 | 23 | 29 | 31 |
| TOF@15 min | 0.56 | 0.62 | 0.85 | 0.98 |
In comparison with the model systems Lbmz1 (TON = 31) and Lpy1 (TON = 22) published earlier, the new systems have an intermediate position (Table 1). There seems to be a clear correlation between TON and TOF; i.e., the faster the catalytic reaction, the higher is the product yield. We assume that a higher rate counteracts side reactions which destroy the catalyst, thus leading to a higher catalytic performance.
The authors would like to thank Deutsche Forschungs-gemeinschaft (DFG), COST and CAU Kiel for financial support as well as Holger Naggert for support with the measurements.
Notes and references
- M. Rolff, J. Schottenheim, H. Decker and F. Tuczek, Chem. Soc. Rev., 2011, 40, 4077 RSC.
- A. W. J. W. Tepper, E. Lonardi, L. Bubacco and G. W. Canters, in Handbook of Metalloproteins, ed. A. Messerschmidt, John Wiley, Chichester, 2010 Search PubMed.
- Á. Sánchez-Ferrer, J. N. Rodríguez-López, F. García-Cánovas and F. García-Carmona, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1995, 1247, 1 CrossRef.
- K. E. van Holde, K. I. Miller and H. Decker, J. Biol. Chem., 2001, 276, 15563 CrossRef CAS PubMed.
- Y. Matoba, T. Kumagai, A. Yamamoto, H. Yoshitsu and M. Sugiyama, J. Biol. Chem., 2006, 281, 8981 CrossRef CAS PubMed.
- H. Decker, T. Schweikardt and F. Tuczek, Angew. Chem., Int. Ed., 2006, 45, 4546 CrossRef CAS PubMed.
- (a) Y. C. Li, Y. Wang, H. B. Jiang and J. P. Deng, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17002 CrossRef CAS PubMed; (b) M. Sendovski, M. Kanteev, V. S. Ben-Yosef, N. Adir and A. Fishman, J. Mol. Biol., 2011, 405, 227 CrossRef CAS PubMed; (c) W. T. Ismaya, H. J. Rozeboom, A. Weijn, J. J. Mes, F. Fusetti, H. J. Wichers and B. W. Dijkstra, Biochemistry, 2011, 50, 5477 CrossRef CAS PubMed.
- K. Selmeczi, M. Réglier, G. Michel and G. Speier, Coord. Chem. Rev., 2003, 245, 191 CrossRef CAS PubMed.
- I. A. Koval, P. Gamez, C. Belle, K. Selmeczi and J. Reedijk, Chem. Soc. Rev., 2006, 35, 814 RSC.
- C. Eicken, C. Gerdemann and B. Krebs, in Handbook of Metalloproteins, ed. A. Messerschmidt, R. Huber, T. Poulos and K. Wieghardt, J. Wiley & Sons, Chichester, UK, 2001, vol. 2, p. 1319 Search PubMed.
- M. Rolff, J. Schottenheim, G. Peters and F. Tuczek, Angew. Chem., Int. Ed., 2010, 49, 6438 CrossRef CAS PubMed.
- (a) K. D. Karlin, S. Kaderli and A. D. Zuberbühler, Acc. Chem. Res., 1997, 30, 139 CrossRef CAS; (b) M. Rolff, J. N. Hamann and F. Tuczek, Angew. Chem., Int. Ed., 2011, 50, 6924 CrossRef CAS PubMed.
- E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047 CrossRef CAS PubMed.
- L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013 CrossRef CAS PubMed.
- A. De, S. Mandal and R. Mukherjee, J. Inorg. Biochem., 2008, 102, 1170 CrossRef CAS PubMed.
- K. D. Karlin, J. C. Hayes, Y. Gultneh, R. W. Cruse, J. W. Mckown, J. P. Hutchinson and J. Zubieta, J. Am. Chem. Soc., 1984, 106, 2121 CrossRef CAS.
- L. Casella, M. Gullotti, M. Bartosek, G. Pallanza and E. Laurenti, J. Chem. Soc., Chem. Commun., 1991, 1235 RSC.
- M. Réglier, C. Jorand and B. Waegell, J. Chem. Soc., Chem. Commun., 1990, 1752 RSC.
- W. Flaig, Th. Ploetz and A. Küllmer, Z. Naturforsch., 1955, 10b, 668 CAS.
- A. Hoffmann, C. Citek, S. Binder, A. Goos, M. Rübhausen, O. Troeppner, I. Ivanović-Burmazović, E. C. Wasinger, T. D. P. Stack and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2013, 52, 5398 CrossRef CAS PubMed.
- J. Schottenheim, N. Fateeva, W. Thimm, J. Krahmer and F. Tuczek, Z. Anorg. Allg. Chem., 2013, 639, 1491 CrossRef CAS.
- K. Kenji, Jp. Pat. 2010-215610, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc47888b |
| This journal is © The Royal Society of Chemistry 2014 |