 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical consequences of mechanical bonding in catenanes and rotaxanes: isomerism, modification, catalysis and molecular machines for synthesis
Edward A.
Neal
and
Stephen M.
Goldup
*
School of Biological and Chemical Science, Queen Mary University of London, Mile End Road, London, E1 4NS, UK. E-mail: s.m.goldup@qmul.ac.uk
First published on 17th January 2014
Abstract
Research on mechanically interlocked molecules has advanced substantially over the last five decades. A large proportion of the published work focusses on the synthesis of these challenging targets, and the subsequent control of the relative position of the covalent sub-components, to generate novel molecular devices and machines. In this Feature Article we instead review some of the less discussed consequences of mechanical bonding for the chemical behaviour of catenanes and rotaxanes, and their application in synthesis, including striking recent examples of molecular machines which carry out complex synthetic tasks.
 Edward A. Neal and Stephen M. Goldup | Steve (right) received his research training at the University of Oxford (MChem, Prof. Sir Jack Baldwin), Imperial College (PhD, Prof. Tony Barrett) and University of Edinburgh (PDRA, Prof. David Leigh). In 2008 he began his independent career at QMUL, where he works with an outstanding group of young scientists developing methods for the synthesis of challenging interlocked molecules with catalytic, materials and sensing applications. Ed (left) joined the Goldup Group as a PhD student in 2011, bringing with him research experience gained at the Universities of Bristol (BSc, Prof. Tom Simpson) and Southampton (MSc, Prof. A. Ganesan). |
1. Introduction
Synthetic methods for the production of mechanically interlocked molecules, in particular catenanes and rotaxanes, have improved immeasurably since the ground-breaking first reports of their synthesis in low yield using statistical methods in the 1960s. These synthetic advances have facilitated the study of mechanically bonded species for a range of applications including electronic devices, sensors, smart materials, and drug delivery systems, as well as their investigation in prototypical molecular machines. Many of these applications, particularly the latter, rely on controlling the relative position of the covalently bonded sub-components of the interlocked structure and stimulating their large-amplitude relative motion between well-defined co-conformations.Applications of catenanes and rotaxanes which rely on other aspects of their unusual chemical properties and, more recently, their utility in organic synthesis both as substrates and catalysts have also been reported. In this Feature Article we briefly introduce catenanes and rotaxanes, and their synthesis before moving on to focus on some of the less discussed effects of mechanical bonding on the physical and chemical properties of interlocked structures. Finally we highlight the relatively recent development of catenanes and rotaxanes for synthetic applications, including exciting examples of molecular machines being employed in chemical synthesis.
2. A brief introduction to catenanes and rotaxanes
Catenanes are mechanically bonded molecules containing two (or more) ring shaped molecules linked together in the manner of links in a chain. Similarly, rotaxanes are composed of one or more rings threaded on one or more dumbbell shaped molecules, where large end groups prevent the macrocycle(s) from escaping. This arrangement ensures that, despite not being linked covalently, they are held together due to the inability of covalent bonds to pass through one another, leading to a mechanical bond. Mechanically bonded molecules are synthetically challenging targets, largely due to the difficulty inherent in trying to arrange two or more independent molecules in space such that a mechanical bond can be formed. Thus, although the first catenane and rotaxane were synthesised respectively by Wasserman in 19601 and Harrison in 1968,2 they remained hard to access curiosities for the majority of the next two decades. Although early directed methods developed by Schill and others led to improvements in the yield of the final mechanical bond forming step, the overall efficiency of the approach was low due to the need for long synthetic routes.3A key breakthrough in the synthesis of mechanically interlocked molecules was provided by Sauvage and co-workers through the demonstration of a non-covalent template method for the synthesis of a [2]catenane,4 in which they employed a tetrahedral CuI ion to organise two phenanthroline ligands, such that when the ends of each ligand were linked, the target catenane was formed in high yield.5 This passive template approach (Fig. 1a), in which the covalent components of the target are held together by a non-covalent “glue” prior to formation of the final covalent bond which establishes the mechanical bond between them, has facilitated developments in the field, and has been extended to a wide range of template motifs and increasingly complicated interlocked structures, often in excellent yield, by Sauvage, Stoddart, Leigh, Vögtle, Harada, Anderson, Beer and others.6
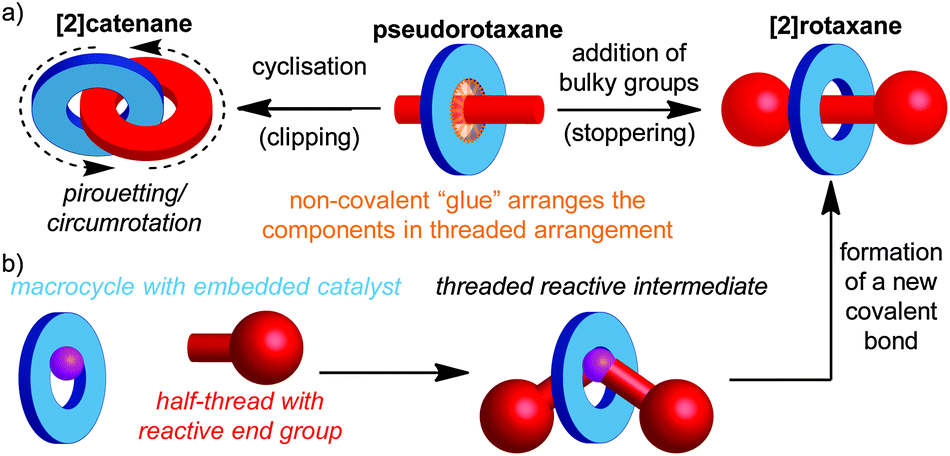 | ||
| Fig. 1 Synthesis of rotaxanes and catenanes by (a) the passive and (b) the active template synthesis of a [2]rotaxane. | ||
More recently, Leigh and co-workers introduced a conceptually novel approach based on an active template, in which a reactive unit in the macrocycle (typically a metal ion) both acts to organise the components and mediate the formation of the mechanical bond. This approach allows the synthesis of rotaxanes (Fig. 1b) and catenanes in excellent yield and can allow sub-stoichiometric quantities of the template to be employed.7
For more details on the synthesis of rotaxanes and catenanes, the interested reader is directed to several excellent recent reviews on methods for the synthesis of mechanically bonded structures.6,7
3. Isomerism in catenanes and rotaxanes
Although the covalent subcomponents of a mechanically bonded molecule are not covalently linked, the mechanical link restricts their relative motion considerably. Most fundamentally, the linear and macrocyclic components in a rotaxane cannot move apart, as the bulky stoppers prevent the macrocycle from escaping the thread and similarly, the rings in a catenane cannot separate, as they cannot pass through one another. Indeed, separation of the covalent subcomponents of a rotaxane or catenane requires the breaking of one or more covalent bonds and thus they are generally considered to be molecules in their own right rather than supramolecular species.8,9As they are independent isomers of their non-interlocked components, rotaxanes and catenanes often display very different physical properties compared with their constituent molecules, including altered solubility,10 photophysical behaviour,11 and optical activity.12 They also display forms of isomerism which are distinct from their non-interlocked counterparts.
3.1 Co-conformational isomerism
The mechanical bond restricts large-amplitude relative motion of the covalent sub-units in rotaxanes to shuttling of the macrocycle along the thread and pirouetting of the macrocycle about the thread axis (Fig. 2). Similarly, in catenanes the rings can rotate with respect to one another and these motions are referred to as pirouetting or circumrotation. For example, in the case of a hetero-[2]catenane, there are two such non-degenerate, large-amplitude motions available (Fig. 1a). Where one ring is significantly larger than the other, circumrotation is often used to describe the motion of the smaller ring around the larger ring and the pirouetting, the rotation of the smaller ring on its own axis.13 Despite these restrictions, in many cases a number of different relative arrangements of the components are possible and these are dubbed co-conformations, in recognition of their similarity with conformations in covalent molecules.14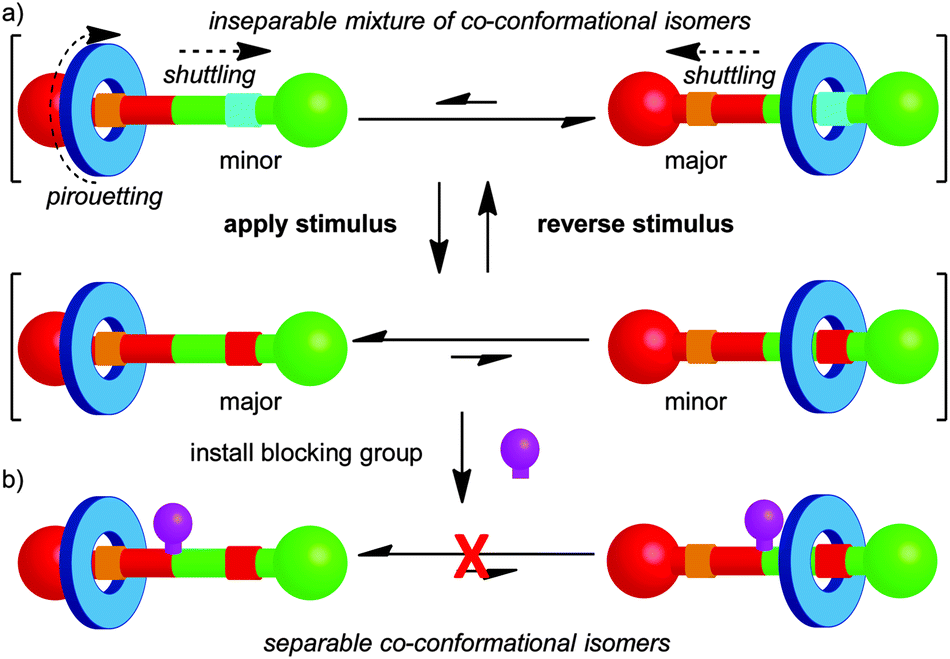 | ||
| Fig. 2 Controlling co-conformation in a [2]rotaxane molecular shuttle using (a) changes in the thermodynamic preference for two binding sites and (b) kinetic barriers to shuttling. | ||
In early examples of catenanes and rotaxanes there were little or no attractive non-covalent interactions between the components.1,2 However, the development of passive template methods allowed the synthesis of catenanes and rotaxanes in which attractive interactions between the covalent components lead to one or more well-defined co-conformation.15 Further, it is possible to alter the co-conformational preference of the system by application of an external stimulus (e.g. acid–base,16 temperature,17 solvent,18 oxidation–reduction,16,19 photochemistry20), which modifies the non-covalent interactions between the components. For example, in a rotaxane it is possible to shuttle the macrocycle from one binding site (termed a station) to the other by application of an appropriate stimulus (Fig. 2). The co-conformational preference of a mechanically interlocked molecule has a large impact on its physical and chemical properties, including fluorescence,21 optical activity,12 conductivity22 and hydrophilicity23 and such molecular shuttles have been applied as sensors,24 smart materials,23 drug delivery vehicles,25 and molecular electronic components.26 This general principle also forms the basis for the development of rotaxanes and catenanes as prototypical molecular machines, as changes in co-conformation lead to large amplitude controlled mechanical motion.27
As with conformational isomerism, when one or more co-conformation is available, the system adopts a Boltzmann distribution between the available states based on their relative free energy. Thus, although there is a large change in the average position of the macrocycle on shuttling, unless the difference in free energy between the two stations is infinite, it is important to note that there will always be a small but finite population of co-conformations where the less favoured binding site is occupied.
Another similarity between conformational and co-conformational isomerism is that, in general, a mixture of co-conformational isomers cannot be separated. However, when the interaction between the components is very strong or exchange between co-conformations is not chemically possible under the reaction conditions, as has been observed in some molecular shuttles based on metal–ligand interactions,28 the co-conformations become separable. Alternatively, it is possible to “trap” the covalent components in a given co-conformation by introducing a steric blocking group that prevents exchange between the available states (Fig. 2b). This approach can be used to ratchet molecular motion,29 and has been applied to great effect by Leigh and co-workers in the development of molecular motors based on [2]catenanes.30
3.2 Connectivity in rotaxanes and catenanes
In their seminal paper on chemical topology, Wasserman and Frisch discussed the potential for isomeric catenanes and rotaxanes with different mechanical connectivity.8 For example, a [3]catenane composed of two different rings can potentially exist as two different isomers (Fig. 3a). Similarly, a [3]rotaxane comprised of a translationally unsymmetrical thread (i.e. the two ends of the thread unit are different) and two different rings can be formed as two different isomers based on the arrangement of the rings on the thread (Fig. 3b). In the case of catenanes these isomers are often referred to as topological isomers as their molecular graphs are non-identical.31 This term does not apply in the case of rotaxanes as both isomers are topologically trivial. However, both cases are reminiscent of constitutional isomers in covalent chemistry, and thus they could be said to differ in their mechanical constitution.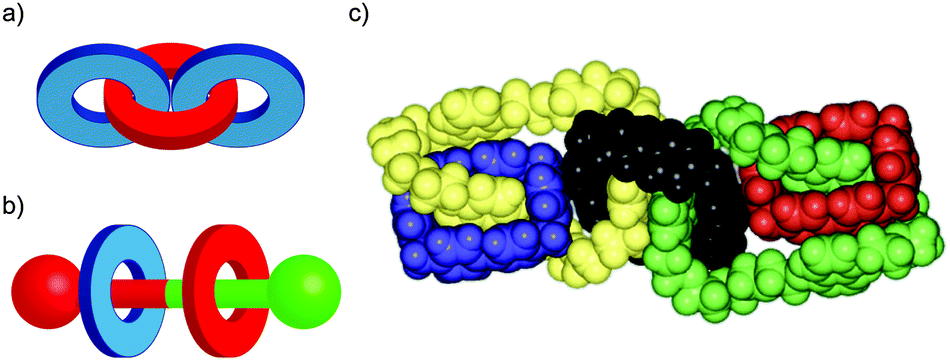 | ||
| Fig. 3 Constitutional isomerism in (a) a [3]catenane; (b) [3]rotaxane; (c) X-ray crystal structure of olympiadane. Fig. 3c from ref. 34 and reproduced with permission from Elsevier. | ||
Although still challenging, methods to selectively synthesise specific isomers of molecules containing multiple mechanically bonded components have been developed, including the selective synthesis of both isomers of a hetero-ring [3]rotaxane by Leigh and co-workers through sequential bond formation,32 and the Stoddart group's impressive synthesis in 1994 of olympiadane, a [5]catenane with the same connectivity as the Olympic logo (Fig. 3c).33,34
3.3 Stereoisomerism of catenanes and rotaxanes
Depending on the molecular symmetry of the sub-components of a mechanically interlocked molecule, there is the potential for stereoisomerism as a consequence of the mechanical bond. For instance, when the thread of a [2]rotaxane is translationally unsymmetrical and the two faces of the macrocycle are differentiated there are two possible diastereoisomers depending on the relative orientation of the components, analogous to the geometrical isomers of alkenes (Fig. 4a). These are separable as they cannot be interconverted without breaking the mechanical bond and examples of their stereoselective synthesis have been reported, particularly in the case of rotaxanes based on cyclodextrin macrocycles.35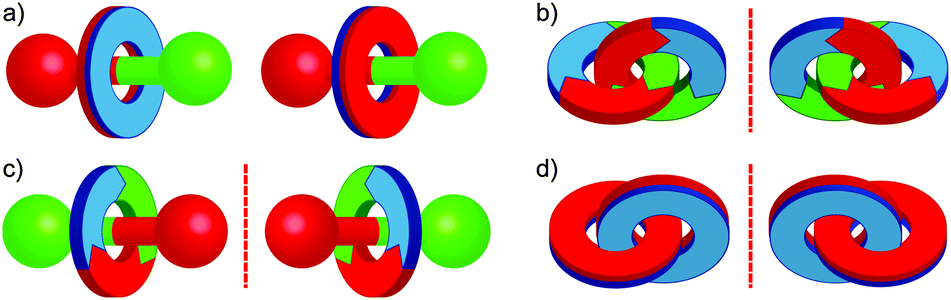 | ||
| Fig. 4 Examples of stereoisomerism in [2]rotaxanes and [2]catenanes: (a) cis and trans isomers; (b) a chiral catenane based on rotationally unsymmetrical macrocycles; (c) a chiral rotaxane composed of a translationally unsymmetrical thread and rotationally unsymmetrical macrocycle; (d) a chiral catenane based on facially unsymmetrical macrocycles. | ||
The formation of the mechanical bond also gives rise to optical isomerism, even when the covalent sub-components themselves are devoid of sources of covalent chirality. Wasserman and Frisch recognised that a [2]catenane composed of two rotationally unsymmetrical rings exists as a pair of mirror images (Fig. 4b).8 Similarly, Schill proposed in 1971 that a [2]rotaxane in which the thread and macrocycle are translationally and rotationally unsymmetrical respectively is a chiral molecule (Fig. 4c).36 In the former case, the catenanes are referred to as topologically chiral as they possess a chiral molecular graph.31 Again, in the case of such chiral rotaxanes, this term does not apply. Given the similar origin of the optical activity, the term mechanically chiral is used below in order to emphasise the structural feature responsible for the optical activity and differentiate sources of covalent chirality from that arising from the mechanically bonded structure.
Although mechanically chiral catenanes and rotaxanes based on rotationally unsymmetrical macrocycles have been synthesised and their enantiomers separated using chiral stationary phase HPLC by Vögtle, Sauvage and others,37 there is currently no published method for the stereoselective synthesis of these challenging targets in enantiopure form,38 which has limited their investigation to date. However, Vögtle and co-workers, using resolved material, demonstrated that mechanically chiral rotaxanes show extremely high molar ellipticity.37 Further, Hiratani and Kameta demonstrated that a racemic mixture of mechanically chiral rotaxanes bind small chiral molecules with high diastereoselectivity.39
It is also possible to generate optically active [2]catenanes if both rings are facially unsymmetrical (Fig. 4d), the first example of which was synthesised as a racemic mixture by Puddephatt and co-workers.40 Later work from Marinetti and co-workers employing a covalently chiral, facially unsymmetrical macrocycle as a single enantiomer allowed the separation of the diastereoisomers of catenane 1 to give both diastereoisomers in optically pure form (Fig. 5).41
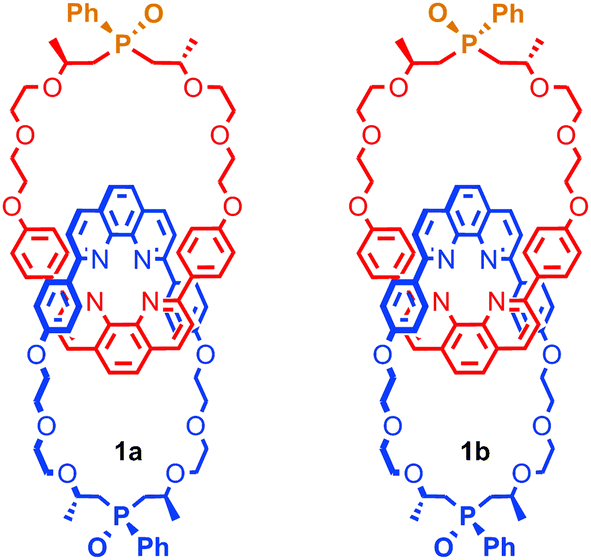 | ||
| Fig. 5 Diastereoisomers of mixed mechanically and covalently chiral [2]catenane 1 synthesised by Marinetti and co-workers and separated using HPLC. | ||
The investigation of stereoisomerism in catenanes and rotaxanes is a relatively underdeveloped area which is ripe for further study. In particular, the development of mechanically chiral catenane and rotaxane catalysts represents an exciting opportunity to apply a new form of molecular asymmetry in the challenging field of enantioselective synthesis. In order to achieve this, novel methods are required to allow stereoselective access to these molecules in enantiopure form.
4. Challenges in the post-synthetic modification of rotaxanes and catenanes – maintaining the mechanical bond
The post synthetic modification of rotaxanes and catenanes by reaction of pendant functionality is relatively trivial, although perhaps underexplored, approach to generating structural diversity in catenanes and rotaxanes. It is also possible to convert a rotaxane into a catenane by cyclisation of the thread component, again an underutilised approach, which allows the synthetic challenges of mechanical bond formation and macrocyclisation to be dealt with separately.42 However, the need to maintain the mechanical bond places restrictions on the modification of key components including the skeleton of the macrocycle and the stoppering units of rotaxanes. A limited number of elegant solutions to these synthetic challenges have been reported.4.1 Replacement of stopper units in rotaxanes
A crucial requirement in any successful post synthetic modification of a rotaxane is that the mechanical bond itself is not disrupted, which places restrictions on the development of successful strategies to replace the stoppering end groups; if stopper is removed the macrocycle is free to escape the thread and the mechanical bond is broken.Stoddart and co-workers developed a grafting approach that allows the stoppering unit of a rotaxane to be replaced without any risk of the system dethreading, by ensuring that all of the intermediates of the reaction are also stoppered. In the first example reported, the bulky phosphonium stopper in rotaxane 2 was replaced using a Wittig reaction; as the bulky phosphorous unit remains associated throughout the process, based on the established mechanism of the Wittig reaction, the steric bulk of the stopper unit is always sufficient to maintain the mechanical bond, even though under the strongly basic conditions of the reaction the crown ether-ammonium template is completely disrupted (Fig. 6a).43 Thus phosphonium rotaxane 2 is converted in high yield into rotaxane 4 where the stopper unit is now an alkene derived from aldehyde 3.
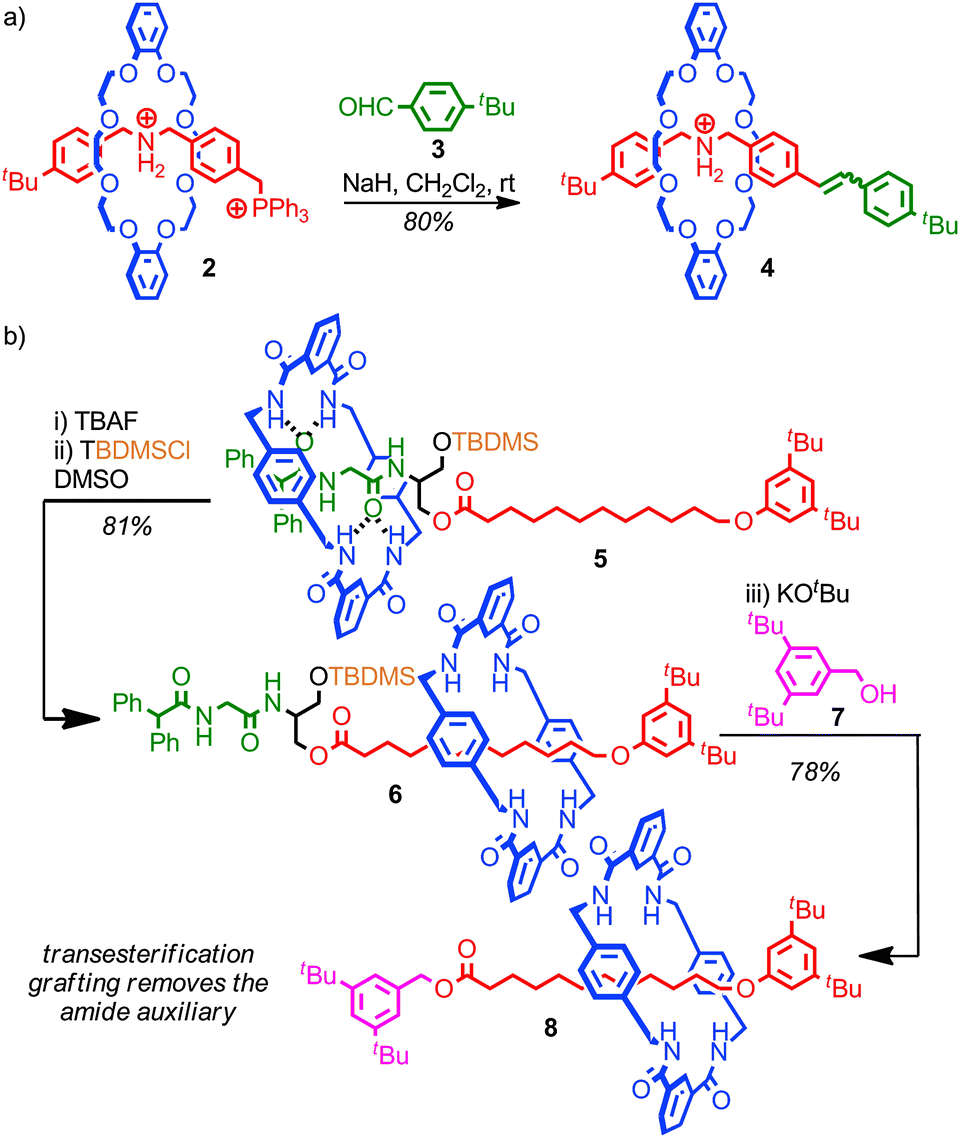 | ||
| Fig. 6 Replacing the stoppers in a [2]rotaxane: (a) Stoddart's original Wittig grafting approach (PF6 anions omitted for clarity); (b) Leigh's mechanical auxiliary synthesis of an “impossible” rotaxane. | ||
The utility of the approach was demonstrated through the synthesis of a [4]rotaxane comprised of three macrocycles around a single thread. This grafting approach has since been extended to more complex structures,44 and other reactions which meet the criteria of producing fully stoppered intermediates including the Tsuji–Trost reaction of allylic ester in which a bulky carbonate stopper is replaced by a substituted malonate,45 transesterification46 and aminolysis47 of simple and activated esters, and the Michael/retro-Michael displacement of vinyl sulfoxides by sulfides.48
Leigh and co-workers extended this concept to produce a mechanically interlocked auxiliary for the synthesis of rotaxanes in which no recognition elements are present, so called “impossible” rotaxanes (Fig. 6b).46 Rotaxane 5 was synthesised through a clipping reaction of the macrocycle around the amide template. The macrocycle is initially confined to the more polar left-hand amide portion of the thread, due to the bulky TBDMS group which acts as a kinetic barrier to shuttling. Subsequent desilylation allows the macrocycle to explore the full length of the thread. In DMSO, the dominant co-conformation is that with the macrocycle on the alkyl right-hand portion of the thread, as this both allows the amide station to hydrogen bond to the DMSO and the macrocycle to shield the lipophilic alkyl chain from the polar solvent. Thus, subsequent resilylation in DMSO yields rotaxane 6 as the major product, in which the macrocycle is now confined to the alkyl portion of the thread. A subsequent transesterification grafting reaction with alcohol 7 yields rotaxane 8 which contains no significant inter-component interactions. Overall, the amide template for the clipping reaction to produce rotaxane 5 acts as an auxiliary for the formation of a mechanical bond between the macrocycle and alkyl thread, which would be extremely challenging to form in any other way.
4.2 Shrinking or expanding the macrocycle
In the same way as replacing the stopper units of a rotaxane runs the risk of breaking the mechanical bond, removing or adding functionality to the skeleton of the macrocycle runs the risk of allowing the covalent sub-components of a rotaxane or catenane to dissociate if the ring is opened during the process. Overcoming this problem in the case of the macrocycle is extremely challenging and to date only a few examples have been reported.Contraction of the ring by removal of a sub-unit has been achieved via the photoextrusion of SO2 from a crown ether sulfone macrocycle (Fig. 7).49,50 Thus, pseudorotaxane 9, in which the macrocycle is large enough to slip over the stopper units (albeit slowly at room temperature) is converted to rotaxane 10 in 28% yield upon irradiation with UV light. The mechanism of the reaction is thought to be the loss of SO2 to give a diradical intermediate, which then recombines to form the new C–C bond. Given that the interlocked product is isolated after ring contraction, it appears that the recombination of the radical chain ends is competitive with the dissociation of the thread and macrocycle.
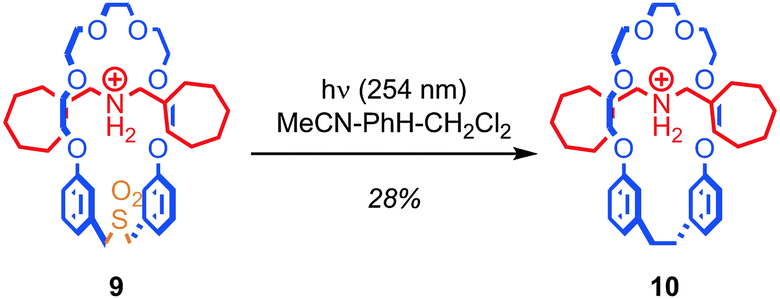 | ||
| Fig. 7 Contraction of a macrocycle via photochemical extrusion of SO2 (PF6 anions omitted for clarity). | ||
Insertion of functionality into the ring skeleton to enlarge the macrocycle is similarly underutilised. Takata and co-workers demonstrated that the Diels–Alder reaction of an exocyclic diene in catenane 11, followed by ozonolysis of the resulting cyclohexene with oxidation of the thioether units, led to catenane 12, in which the macrocycle skeleton has been expanded by six atoms in reasonable yield (Fig. 8a).51 Recently, Advincula and co-workers demonstrated that a living ring-opening polymerisation can be used to expand the macrocycles of [2]catenane 13 which contain reactive stannyl ester units, to deliver catenated cyclic polymers 14 that the authors successfully visualised using AFM on a mica surface.52
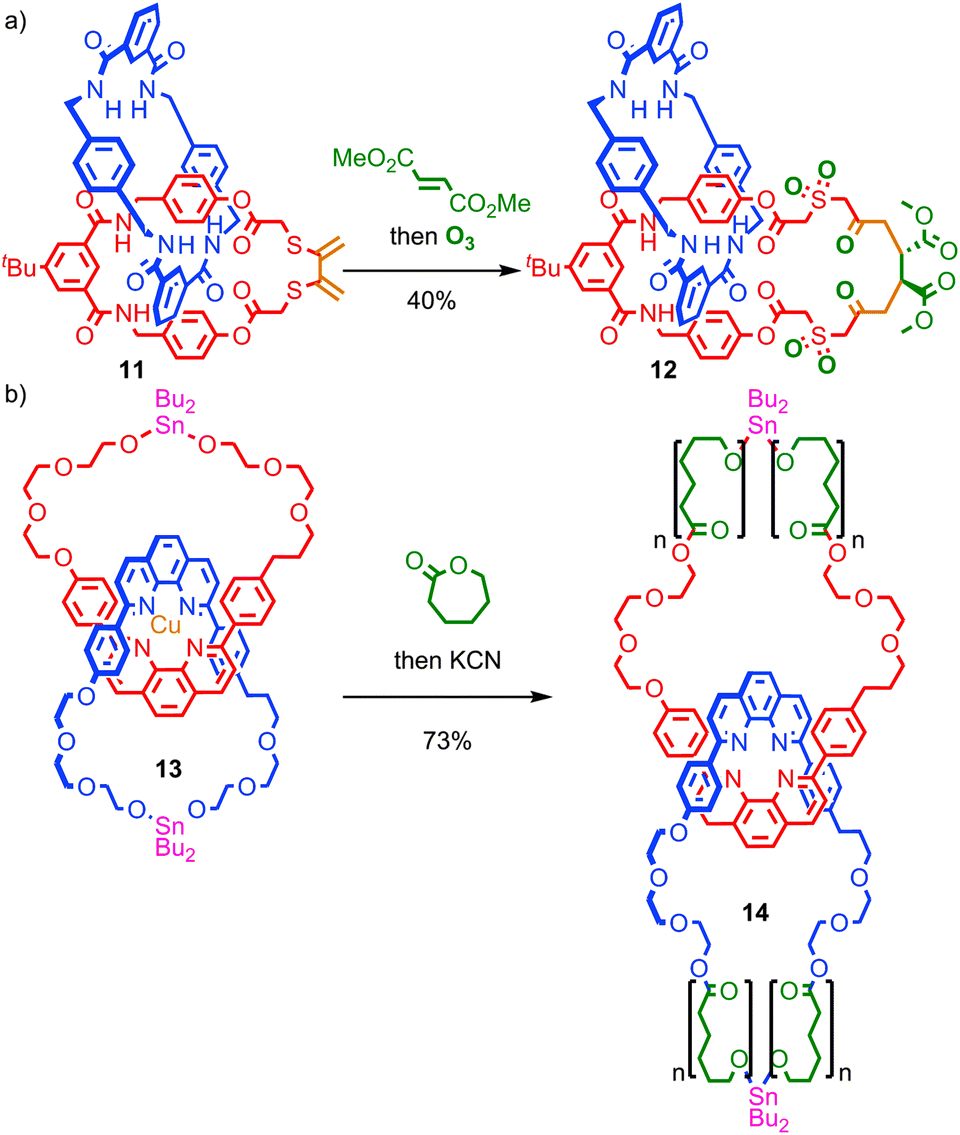 | ||
| Fig. 8 Expansion of macrocycles in [2]catenanes: (a) a Diels–Alder/ozonolysis approach developed by Takata et al.; (b) Advincula and co-workers' living ring opening polymerisation of [2]catenane 13 to deliver catenated polymers 14. | ||
5. The effect of mechanical bonding and chemical reactivity
The mechanical bond alters the chemistry of catenanes and rotaxanes by restricting the relative motion of the subcomponents, increasing steric hindrance of reactive functionalities and altering the strength of intercomponent non-covalent interactions with important implications for their chemistry and applications.5.1 The effect of mechanical bonding on non-covalent interactions
Perhaps the most obvious effect of mechanical bonding is to increase the strength of non-covalent interactions between the components. This became apparent early in the development of passive template methods with the observation by Sauvage and co-workers that the removal of the CuI template from the [2]catenane product required much more harsh conditions than those required to demetallate a simple homoleptic CuI-phenanthroline complex, for which the authors coined the term “catenand effect”.53 Similarly, the pKa of the ammonium unit in rotaxanes synthesised using a crown ether-ammonium template is significantly increased compared to a mixture of the non-interlocked components.54This strengthening of the interaction between components is attributed to the loss of degrees of freedom, due to the formation of mechanical bond. In keeping with this, the effect is larger for systems where conformational and/or co-conformational flexibility is minimised. Thus in rotaxanes 15, in which the macrocycle size was systematically varied, the C–H hydrogen bond between the triazole proton of the thread and the bipyridine nitrogen of the macrocycle increases in strength (as judged by the chemical shift of the triazole proton in 1H NMR) as the macrocycle size is reduced (Fig. 9).55 A similar effect was observed with variation in thread length; shorter threads led to a stronger C–H hydrogen bond, with the shortest thread examined giving a rotaxane in which the triazole C–H resonated at 10.5 ppm, an approximately 2.5 ppm shift relative to the non-interlocked thread.
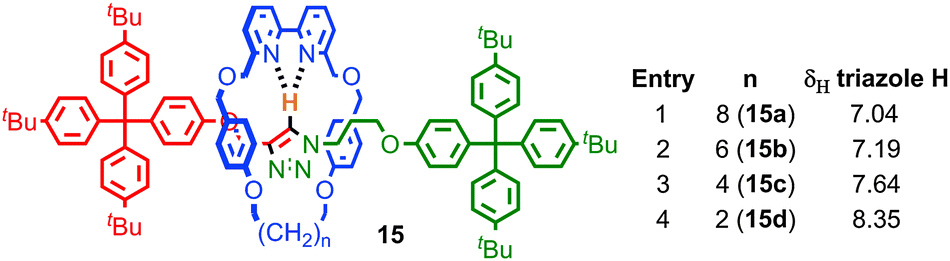 | ||
| Fig. 9 Effect of macrocycle size on the strength of a C–H hydrogen bond as measured by the 1H NMR chemical shift of the triazole C–H. | ||
5.2 The effect of mechanical bonding on chemical reactivity
The mechanical bond can also play a large role in reducing the kinetic reactivity of the products as demonstrated by Vögtle and co-workers: the hydrogenation of the alkene in the thread of rotaxane 16 to give rotaxane 17 was significantly retarded compared to the non-interlocked thread, with significantly lower yields of the product observed over the same reaction time (Fig. 10).56 The reduction in reactivity was ascribed to the steric hindrance caused by the mechanical bond. A similar effect was observed by Leigh and co-workers in the Diels–Alder reaction of a fumaramide unit in the thread of a rotaxane and by Prato and Mateo-Alonso in the stabilisation of fulleropyrrolidine N-oxides.57,58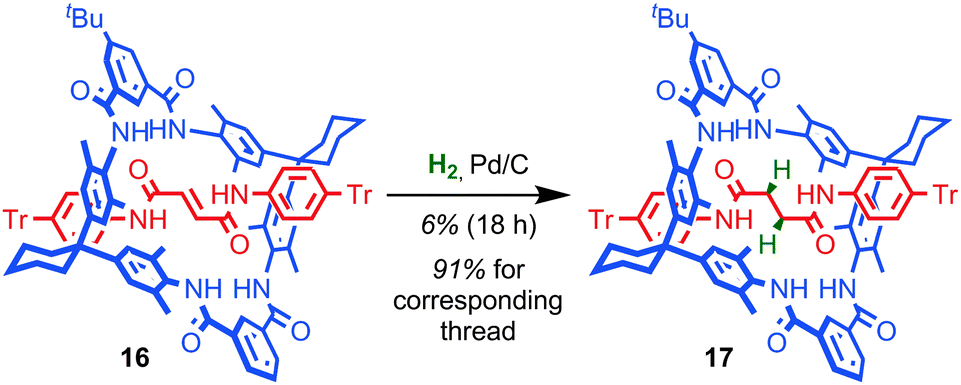 | ||
| Fig. 10 The effect of the mechanical bond on the reactivity of an alkene towards hydrogenation. | ||
The steric hindrance of the rotaxane architecture has been shown to kinetically stabilise otherwise reactive intermediates and allow their isolation. Takata and co-workers reported the unexpected isolation and characterisation of thiophosphonium rotaxane 19, a reactive intermediate of phosphine-mediated cleavage of disulfide rotaxane 18 (Fig. 11a).59 The equivalent reaction of the non-interlocked thread did not lead to an isolable thiophosphonium salt. Recently, Goldup and co-workers serendipitously isolated CuI-triazolide rotaxane 20, the intermediate of an active template Cu-mediated azide–alkyne cycloaddition reaction, in which the normally highly reactive Cu–C bond is protected by the mechanical bond (Fig. 11b).60 Even carboxylic acids cleave the Cu–C bond only slowly to cleanly produce triazole rotaxane 21 and it is even possible to isolate triazolide 20 in excellent yield under aqueous conditions.
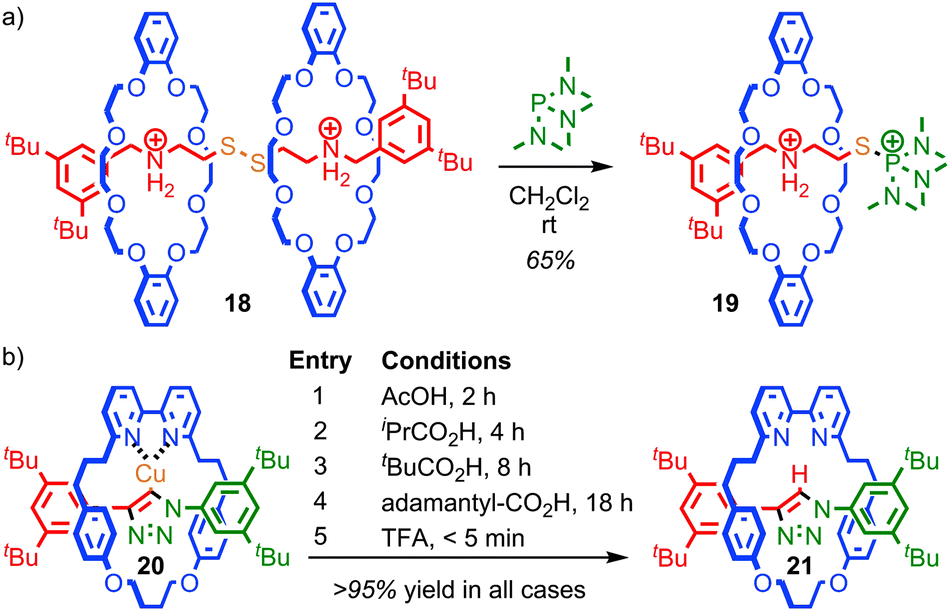 | ||
| Fig. 11 Mechanical stabilisation reactive intermediates: (a) thiophosphonium rotaxane 19 (PF6 anions omitted for clarity) isolated by Takata et al.; (b) the CuI-triazolide rotaxane isolated by Goldup and co-workers. | ||
In contrast, the interaction between subcomponents can also serve to increase the reactivity of functionality through non-covalent activation. An interesting example which combines kinetic deactivation and thermodynamic activation was reported by Leigh and co-workers.61 The authors found that the nitrone functionality of rotaxane 22 was protected from reduction by small chemical reductants such as NaBH4 by the mechanical bond; rotaxane 22 was recovered unchanged after 12 h while corresponding thread was reduced in quantitative yield in just 30 min under the same conditions (Fig. 12). Conversely, cyclic voltammetry revealed that the first reduction wave of the nitrone group in rotaxane 22 occurs at E1/2 = −1.3 V compared with E1/2 = −1.7 V for the non-interlocked thread, indicating that the first step in the reduction is ∼8 kcal mol−1 more favourable. However, a kinetic study revealed that although more favourable, the standard rate constant for the single electron reduction was two orders of magnitude lower for the rotaxane than for the non-interlocked thread. Thus it appears that the system is kinetically deactivated but electronically activated towards reduction.
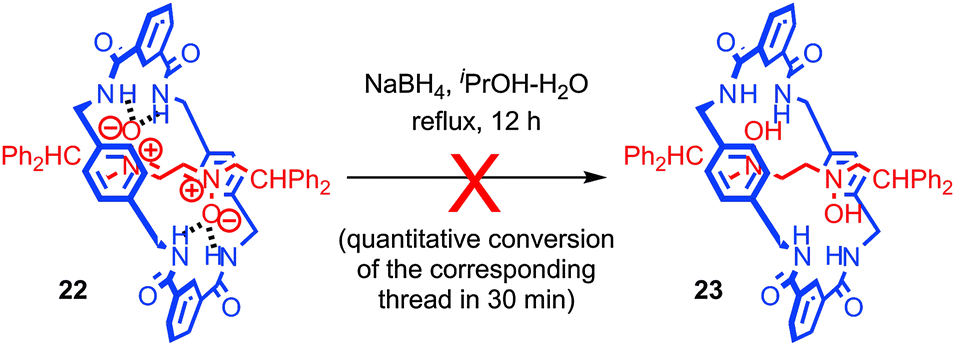 | ||
| Fig. 12 Sterically deactivated but electronically activated nitrone rotaxane 22. | ||
It is clear from the examples discussed, that mechanical bonding has a large effect on the intrinsic chemistry of the functional groups present in the covalent subcomponents of catenanes and rotaxanes. This potential to modify reactivity without altering the covalent structure and thus maintain other desirable properties has led to a catenanes and rotaxanes being investigated for a number of technological applications.
5.3 Applications of mechanical bonding in controlling reactivity
The tendency of the mechanical bond to alter the reactivity of the components, in particular to protect against unwanted reactions, has been exploited in a number of areas. One of the first applications was in the protection of organic dyes from environmental degradation: Anderson and co-workers demonstrated that organic dyes such as azo- and cyanine-dyes could be encapsulated in a rotaxane architecture and that this encapsulation enhanced their stability to photobleaching and other environmental degradation pathways, as well as enhancing their fluorescence.62 Later, Smith and co-workers demonstrated that squaraine dyes, which are unstable under biologically-relevant conditions, but highly fluorescent in the deep red and near infrared (NIR) region – a highly desirable property for biological applications – could be stabilised in a rotaxane architecture.63Anderson and co-workers demonstrated that conjugated polymers threaded in cyclodextrin macrocycles to form a polyrotaxane insulated molecular wires (IMWs), such as polyvinylphenylene rotaxane 24, display enhanced stability compared with their non-interlocked analogues (Fig. 13).64 The insulation of the molecular wire in a rotaxane architecture also prevents aggregation of the conjugated polymers in the solid state and reduces the effect of external fluorescence-quenching species; the size of the macrocycle strongly influences the effect of insulation, enhancing their photophysical and electro-luminescent properties for device applications (Fig. 13). Rotaxane IMWs have been proposed as components of plastic electronic devices such as OLEDS, TFTs and organic solid state lasers.65 This approach has since been extended to other insulated oligomeric and polymeric electronic materials by Anderson and others, including the radical cationic form of an oligomer of polyaniline.66
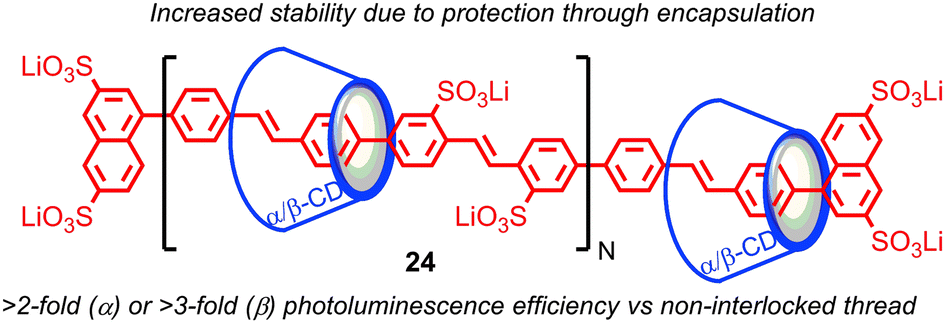 | ||
| Fig. 13 Enhancement of the photoluminescent properties of a polyphenylene molecular wire in a polyrotaxane architecture. | ||
Stoddart and co-workers have developed a radical π-mer template for the synthesis of rotaxanes and catenanes that has allowed them to synthesise and study mechanically bonded organic radicals based on the viologen radical cation. Impressively, this approach allowed them to access [2]catenane 25 which displays a remarkable six oxidation states.67 Under ambient conditions catenane 25 exists as a mixture of the +7 (monoradical) and +6 (diradical) oxidation states (Fig. 14), the closed shell +8 state being disfavoured due to high coulombic repulsion. Chemical oxidation with tris(4-bromophenyl) aminium hexachloroantimonate allowed the +8 state to be characterised fully by NMR. Square-wave differential pulsed voltammetry allowed the closed shell +8, and open shell +7, +6, +4, +2 and +0 species to be accessed reversibly, allowing the system to be switched between multiple oxidation states. When the stability of a series of analogous rotaxanes was studied, the stability of the reduced species was found to depend on the length of the rotaxane thread indicating that the highly restricted nature of the mechanical bond is key in stabilising the normally reactive paraquat radical cation.68
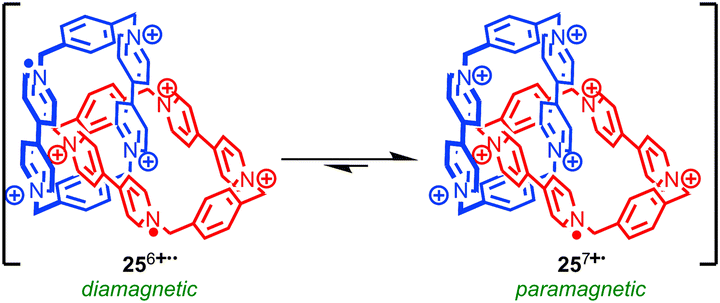 | ||
| Fig. 14 Stoddart's radically configurable [2]catenane (PF6 anions omitted for clarity). | ||
Papot, Aucagne, Leigh and co-workers demonstrated a twofold role for the steric protection of bioactive peptide, met-enkephalin (also known as opiate growth factor, OPG), in a rotaxane architecture in order to develop a prototypical rotaxane pro-drug.69,70 Peptidic drugs are exciting therapeutic candidates, but their stability in vivo is often poor. Macrocycle 28 wrapped around the peptide thread of rotaxane 26 prevents the degradation of OPG by peptidases and also leaves it in a biologically inactive form. Cleavage of the stopper unit by the enzyme β-galactosidase releases active peptide 27, suggesting that this approach could be used to deliver peptide drugs and allow their targeted release (Fig. 15). This is particularly useful in the case of OPG as it possesses both anti-tumour activity and influences perception of pain. Thus it would be extremely advantageous to be able to administer it in an inactive form and target its activation to the tissue of interest. In a follow up study the authors enhance the approach further by equipping the macrocycle with solubilising groups, increasing the rotaxane's solubility by four orders of magnitude relative to the corresponding thread, thereby addressing another key problem experienced in the development of peptidic drugs, bioavailability.71
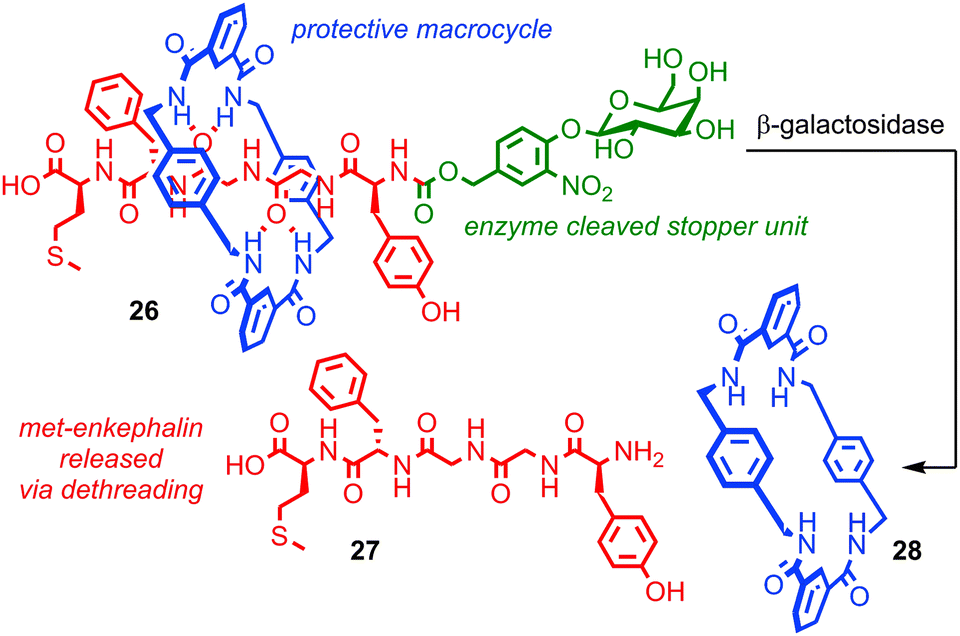 | ||
| Fig. 15 A rotaxane pro-peptide for the targeted delivery of bio-active peptide 27. β-Galactosidase cleaves the glycosidic bond which removes the stopper group and allows the macrocycle to escape, liberating the peptide. | ||
In 2001, Okumuro and Ito demonstrated a very different role for mechanical bonding in controlling the mechano-chemistry of polymers by introducing mobile cross links between the polymer chains based on cyclodextrin macrocycles (Fig. 16a).72,73 Here the purpose of the mechanical bond is not to enhance or reduce reactivity per se, but to allow the strain experienced by a polymer under deformation to be distributed over the network in a manner akin to a pulley in a network of cables (Fig. 16b) and thus reduce the driving force for mechanically induced chain scission. These slide-ring polymers and gel show dramatically enhanced tensile strength among other unusual properties, such as remarkable swelling properties (Fig. 16c), and have begun to find application in scratch resistant coatings and paints.
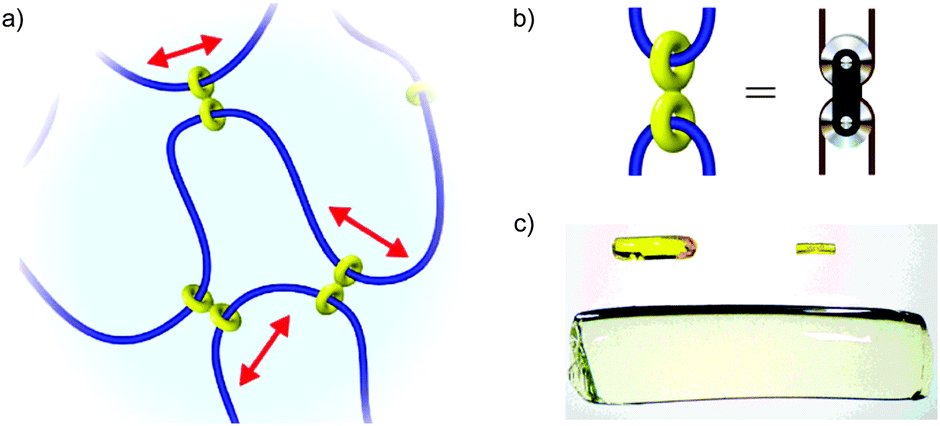 | ||
| Fig. 16 Slide ring polymers: (a) schematic diagram of a slide ring cross-linked polymer; (b) the figure of eight cross link acts like a macroscopic pulley to distribute stress over the polymer network; (c) example of the volume change possible in a polyrotaxane gel: left: as synthesised (65 mg). Right: the dry gel (7.2 mg). Below: the water-swollen gel (2.93 g). The polyrotaxane gel absorbs water up to ca. 400 times its dry weight (reproduced with permission from Adv. Mater.). Fig. 16a and b from ref. 72 and reproduced with permission from Nature Publishing Group. Fig. 16c from ref. 73 and reproduced with permission from John Wiley and Sons. | ||
Recent work from the Smith group has combined the stabilisation of a squaraine dye in a rotaxane architecture with control over chemical reactivity to generate chemoluminescent NIR biosensors (Fig. 17).74 Irradiation of rotaxane 29 with red light in the presence of O2 led selectively to mono-endoperoxide rotaxane 30 which is stable indefinitely at −20 °C. On warming to 37 °C the endoperoxide undergoes smooth cycloreversion to regenerate rotaxane 29 and singlet oxygen, with concomitant excitation of the squaraine fluorophore leading to chemoluminescence in the NIR. Rotaxane 30 was investigated in vivo as a prototypical chemoluminescent biosensor. The behaviour of rotaxane 29 is noteworthy in that (i) the parent macrocycle is known to undergo cycloaddition with singlet oxygen on both anthracene units to generate the corresponding di-endoperoxide and (ii) the thermal decomposition of endoperoxides typically requires higher temperatures and leads to skeletal rearrangement rather than smooth cycloreversion.75 Thus it appears that the rotaxane architecture is serving both to control the initial cycloaddition and sterically activate the cycloreversion process while directing the reaction to give quantitative recovery of the rotaxane starting material.
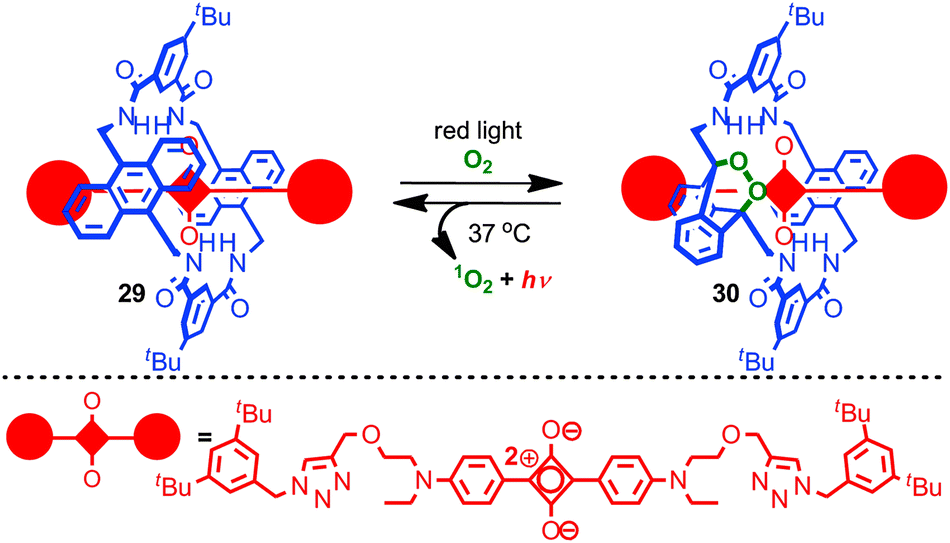 | ||
| Fig. 17 Smith's NIR chemoluminescent rotaxane. Irradiation of rotaxane 29 with red light leads to formation of endoperoxide 30 which can be stored at −20 °C. 30 undergoes cycloreversion at 37 °C to produce 1O2 and regenerate rotaxane 29. | ||
Finally, Leigh and co-workers have demonstrated a very different role for mechanical bonding in controlling chemical reactivity in which the position of the macrocycle in a molecular shuttle determines the rate of a chemical reaction (Fig. 18). The non-interlocked thread of rotaxane 31 is achiral. However, the two co-conformations of rotaxane 31 are chiral, as the position of the macrocycle desymmetrises the thread, rendering the central carbon bearing the hydroxyl group stereogenic. These enantiomeric co-conformations exist as a racemic mixture and react with benzoic anhydride and DMAP at an equal rate to produce racemic rotaxane 33, in which the macrocycle is now prevented from exploring the thread by the bulky benzoyl ester. However, when chiral acylation catalyst (S)-32 is employed, a dynamic kinetic resolution takes place: the two enantiomeric co-conformations of 31 react at different rates and produce an enantioenriched mixture of rotaxanes 33 (34% ee). When the opposite enantiomer of catalyst 32 is employed, the opposite enantiomer of rotaxane 33 dominates in the product mixture. In molecular machine terms rotaxane 32 is operating as an information ratchet,76 and this principal has since been extended to a 3-compartment prototypical linear molecular motor.
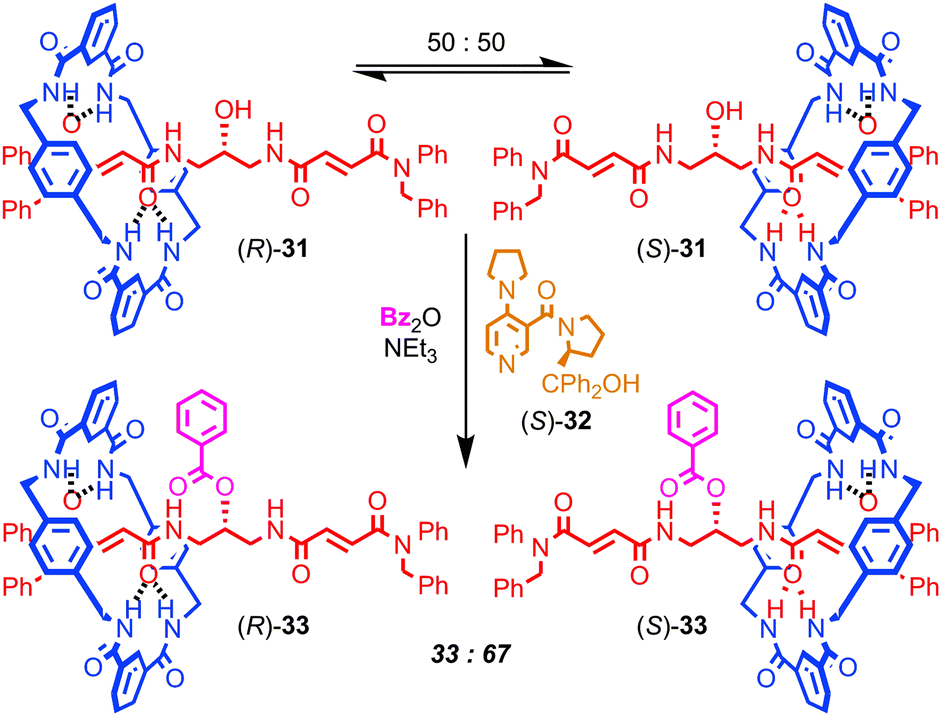 | ||
| Fig. 18 A chemically driven information ratchet. Dynamic kinetic resolution of the enantiomeric co-conformations of [2]rotaxane 31 leads to displacement of the macrocycle. | ||
6. Rotaxanes and catenanes as reagents and catalysts
Given the extensive use of catalytically active metals such as Cu and Pd, as well as organocatalytic functionality such as amines, in the construction of catenanes and rotaxanes, it is perhaps surprising that very few examples in which these residual functionalities are exploited in the development of mechanically interlocked catalysts have been reported. An obvious reason for this apparent omission is that catenanes and rotaxanes produced via passive template methods typically give rise to coordinatively saturated transition metal complexes that are poorly suited to catalysis.The first example of a catalytically active rotaxane, thiazolium-containing rotaxane 34, was reported by Takata and co-workers in 2004 (Fig. 19). The chiral binaphthyl unit in the macrocycle allowed the thiazolium unit in the thread to mediate a benzoin reaction in good yield and reasonable enantioselectivity. This result demonstrates not only that the mechanical bond can provide an unusual, sterically crowded reaction field for catalysis, but also that chiral information transfer is possible from one component (here the macrocycle) to a catalytic functionality in the other (the thread).
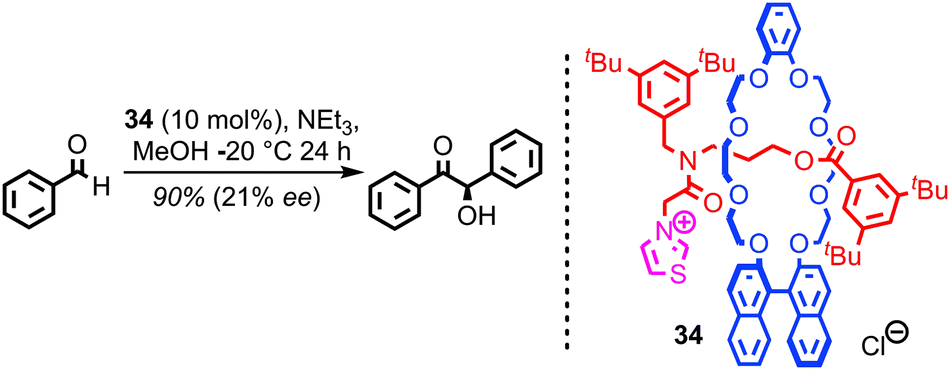 | ||
| Fig. 19 Takata's chiral rotaxane organocatalyst 34. The chiral information in the macrocycle is able to induce enantioselectivity in a benzoin condensation mediated by the thread. | ||
With regards to transition metal catalysis, Hagiwara and co-workers have reported a bis-phosphine containing [2]catenane as a ligand for Pd in a simple Suzuki coupling. However, no comparison was provided between the interlocked structure and its non-interlocked analogue to demonstrate the advantages of the catenane over two equivalents of the corresponding macrocycle.77 In 2007 and 2008 respectively, Nishibayashi and Fan independently reported chiral [2]pseudorotaxane rhodium complexes as enantioselective hydrogenation catalysts.78,79 Although not permanently interlocked, these systems demonstrate the potential of threaded architectures in transition metal catalysis; [2]pseudorotaxane 35 was shown by Nishibayashi et al. to provide superior enantioselectivity and activity in the hydrogenation of enamides than the chiral macrocycle alone (Fig. 20). The authors also demonstrated the inherent modularity of their approach by variation of the thread and macrocycle structure, which led to large variations in the efficiency and selectivity of the reaction.80
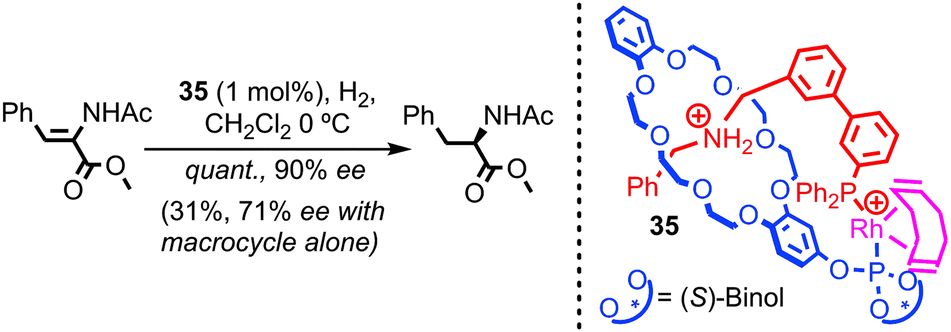 | ||
| Fig. 20 Nishibayashi's [2]pseudorotaxane enantioselective hydrogenation catalyst. Pseudorotaxane 35 displays superior reactivity and selectivity compared with the macrocycle alone (PF6 anions omitted for clarity). | ||
Loeb, Stephan and co-workers used the steric protection of a Lewis basic amine by the mechanical bond to develop a novel frustrated Lewis base for the activation of hydrogen.81 When rotaxane 36 and a boron Lewis acid were treated with H2, protonated rotaxane 37 and the corresponding borohydride anion were produced, resulting from the heterolytic activation of hydrogen (Fig. 21). The steric hindrance of the mechanical bond is essential for this chemistry; when the corresponding non-interlocked thread was employed, no activation of H2 was observed. Given the general interest in frustrated Lewis pairs in the development of novel reactions and reactivity, this application of the unusual, sterically crowded environment of the mechanical bond seems a promising approach.
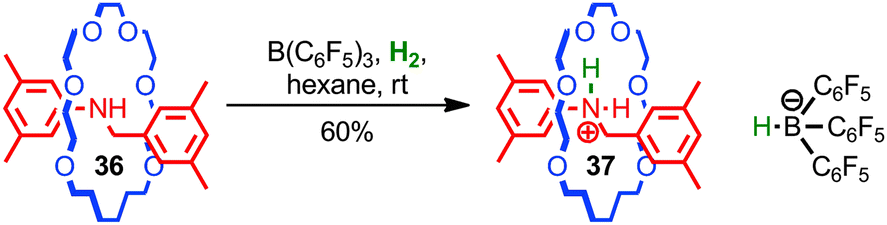 | ||
| Fig. 21 Loeb's [2]rotaxane frustrated Lewis base: in combination with B(C6H5)3 rotaxane 36 achieves the heterolytic activation of H2 at room temperature. | ||
A recent report from Berná and co-workers demonstrated the use of an azodicarboxamide-containing molecular shuttle in a catalytic Mitsonobu esterification (Fig. 22).82 Stoichiometric studies demonstrated that the reaction of PPh3 with rotaxane 38 to give phosphonium rotaxane 39 takes place with concomitant shuttling of the macrocycle from the azodicarboxamide station to the succinamide station. Rotaxane 39 then reacts with an aliphatic alcohol to give reduced rotaxane 40, in which the major co-conformation is still that in which the macrocycle encircles the succinamide station. Re-oxidation of the rotaxane 40 by iodosobenzene diacetate regenerates rotaxane 38 and the macrocycle then returns to the azodicarboxamide station. Thus, when rotaxane 38 was employed sub-stoichiometrically, the catalytic cycle proceeds with shuttling of the macrocycle between the two stations. The authors also demonstrated that the binding of the macrocycle to the azodicarboximide station activated the azo functionality to attack by a phosphine, presumably due to the Lewis acidic effect of the hydrogen bond between macrocycle N–H and the thread carbonyl.
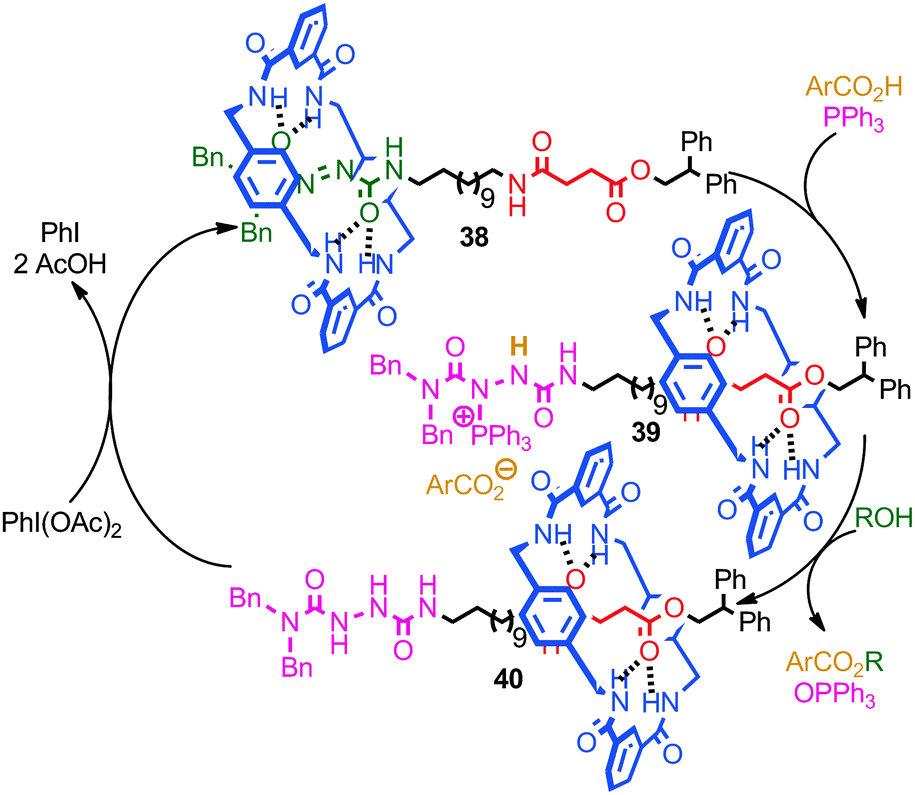 | ||
| Fig. 22 Berná's catalytic azodicarboxamide rotaxane for the Mitsunobu esterification of alcohols. Stoichiometric studies demonstrated that the macrocycle shuttles between the two stations during the catalytic cycle. | ||
Osakada and co-workers have developed a di-nuclear Pd catalyst based on a [3]rotaxane architecture which mediates the Mizoroki–Heck reaction.83 The purpose of the rotaxane architecture is to provide a flexible scaffold, arranging the two palladium centres a suitable distance apart to encourage macrocyclisation over oligomerisation when a bifunctional alkene and organohalide are employed (Fig. 23). The authors demonstrated that the yield of macrocycle 43 produced with rotaxane catalyst 44 is slightly enhanced relative to using a simple mono-nuclear Pd catalyst, although it should be noted that the effect of the rotaxane architecture appears small and that the comparison between the 44 and Pd(PPh3)2(OAc)2 is perhaps uninformative, given the dramatically different ligands bound to the Pd centre.
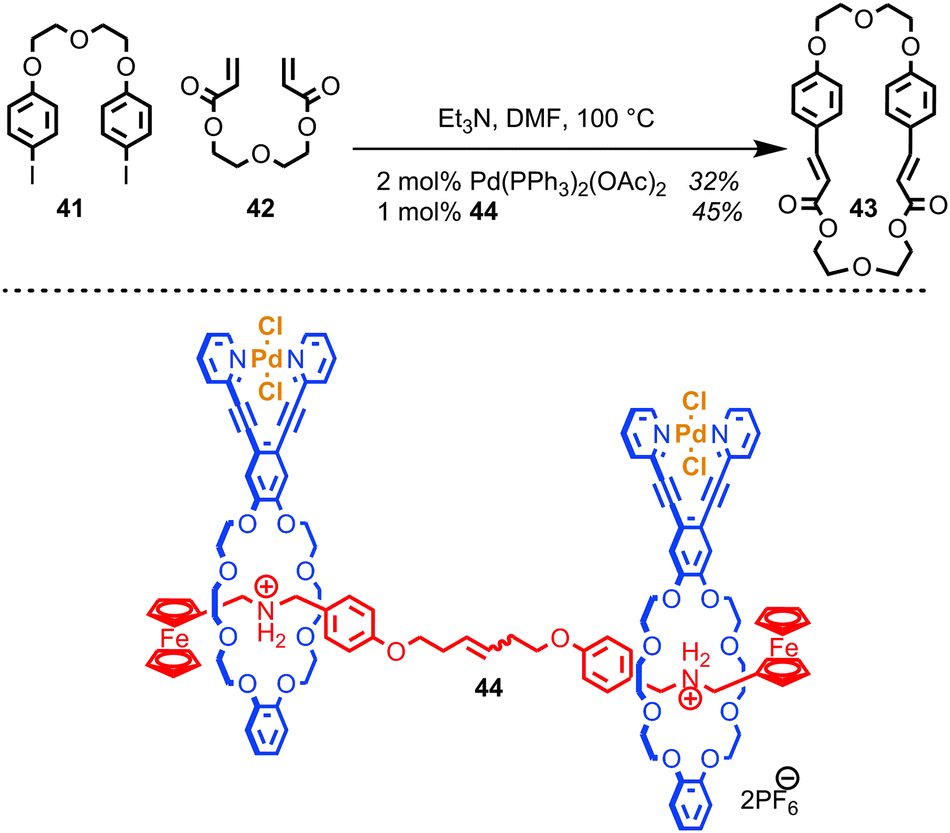 | ||
| Fig. 23 Osakada's [3]rotaxane Pd catalyst for the Mizoroki–Heck reaction. | ||
Leigh and co-workers recently reported a rotaxane catalyst which mimics one of the quintessential functions of an enzyme: the ability to regulate its activity in response to external stimuli.84 They achieved this by employing the steric hindrance provided by the macrocycle to reversibly cover and reveal a catalytic site using large-amplitude molecular motion, much as the regulation of enzymes is often achieved by opening and closing channels which give access to the active site. The system reported is based on the application of non-degenerate crown ether-based molecular shuttle as an organo-catalyst for the conjugate addition of a thiol to cinnamaldehyde. When the central amine unit is protonated it is encircled by the macrocycle and rotaxane 45 demonstrates no catalytic activity over 5 days at room temperature (Fig. 24). In contrast, the non-interlocked ammonium thread achieves 49% conversion under the same conditions. However, when the ammonium unit in rotaxane 45 is deprotonated to give rotaxane 46 simply by shaking a solution of 45 with NaOH, the macrocycle occupies the triazolium stations, revealing the organocatalytic amine moiety. Rotaxane 46 catalyses the conjugate addition reaction efficiently, achieving 83% conversion over 5 days at room temperature.
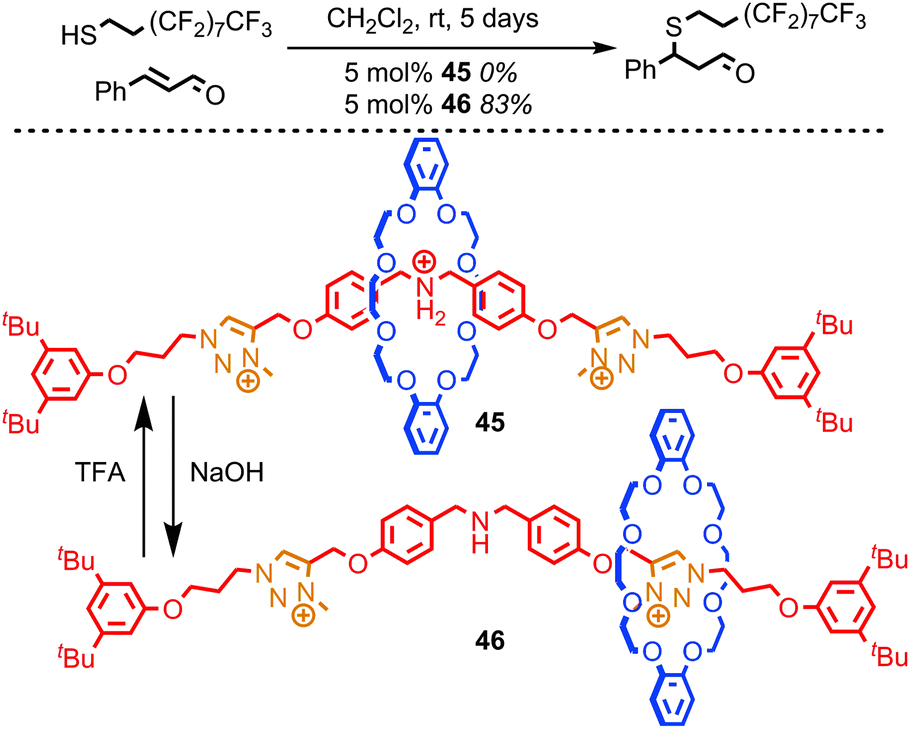 | ||
| Fig. 24 Leigh's switchable organocatalytic rotaxane for the thiol-Michael addition (PF6 anions omitted for clarity). Switching of the co-conformation through protonation–deprotonation of the central amine unit switches the catalytic activity on and off. | ||
7. Processive rotaxane catalysts – towards molecular machines for synthesis
The replication, synthesis and degradation of biopolymers in living cells are carried out by enzymes that are remarkable for their processivity (the number of steps they take before the enzyme and template dissociate) and accuracy. In the case of many DNA polymerase and exonuclease enzymes, these properties are a consequence of the unusual arrangement of enzyme and substrate: the substrate threads through a ring-shaped portion of the enzyme termed the sliding clamp; this mechanical bond holds template and catalyst together, minimising the risk of them moving apart again before the operation is complete.85 This mechanical link confers a significant increase in reaction rate, as well as dramatically increasing the processivity compared with the non-threaded architecture.86,87 Recently, a number of groups have reported non-natural molecular devices that mimic aspects of the behaviour of these complex biological molecular machines.In 2003, Rowan and Nolte reported the first example of an artificial processive molecular machine by threading a polybutadiene unit through a urea-clip macrocycle containing a catalytically active manganese porphyrin to form a threaded structure (Fig. 25a).88,89 Capping of the porphyrin with a bulky pyridine ligand prevented the catalyst from acting on substrates not threaded through the cavity. Subsequent addition of tBuOOH allowed the macrocycle to epoxidise the butadiene thread rapidly. Like the natural molecular machine it aimed to mimic, the epoxidation reaction is highly processive (the catalyst and substrate remain associated over multiple rounds of catalysis), with the unusual catalyst–substrate architecture accelerating the reaction dramatically. Further, as a consequence of the unusual catalyst–substrate arrangement the stereochemistry of the epoxide product (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 cis–trans) is different to that produced by a non-interlocked Mn–porphyrin catalyst (20:80 cis–trans).
:20 cis–trans) is different to that produced by a non-interlocked Mn–porphyrin catalyst (20:80 cis–trans).
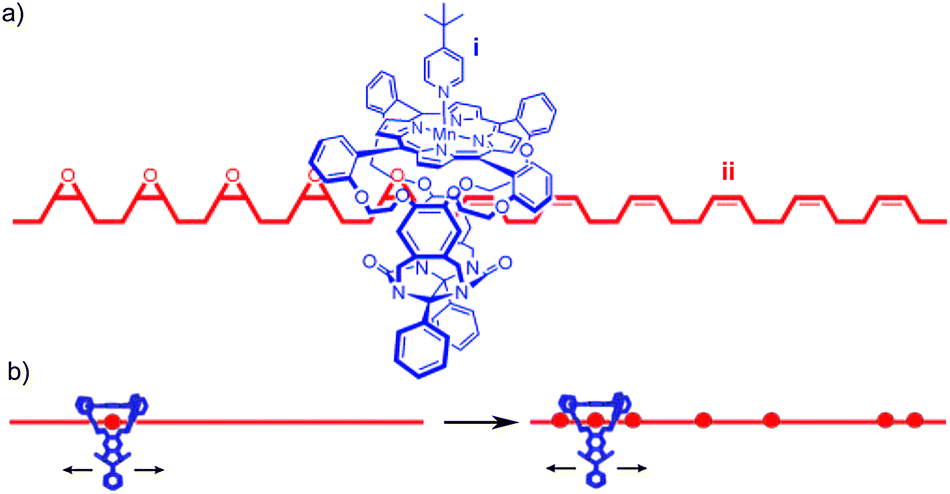 | ||
| Fig. 25 Rowan and Nolte's DNA polymerase mimic: (a) fragment of rotaxane structure showing (i) macrocycle containing the Mn porphyrin catalyst and (ii) the partially oxidised polybutadiene thread resulting from the initially proposed sequential oxidation process; (b) a schematic of the now accepted random walk oxidation process. From ref. 89 and reprinted with permission from the National Academy of Sciences U. S. A. | ||
Originally the reaction was thought to take place in sequence, with each double bond transformed into the epoxide product in turn, as shown in Fig. 25a. In subsequent publications, the authors reported detailed and elegant investigations of the mechanism of polymer threading through the cavity of the macrocycle and the rate of macrocycle diffusion along the polybutadiene thread. Ultimately it was determined that, although the epoxidation of polybutadiene by such catalysts is highly processive, it does not happen in sequence; the macrocycle diffuses along the polymer far more rapidly than the rate of catalyst turn over and thus randomly epoxidises the reactive alkene units (Fig. 25b).88 Takata and co-workers reported a similar result in the cyclo-isomerisation reaction of a thread containing allylic carbamates mediated by a macrocyclic Pd catalyst.90
In a recent publication, Rowan and Nolte report an impressive bio-hybrid catalyst which combines a naturally occurring DNA clamp from the bacteriophage T4 replisome with a small molecule catalyst.91 The authors succeeded in substituting the clamp enzyme with three equivalents of a Mn porphyrin catalyst that is able to oxidise DNA. By running the reaction both with and without an octapeptide, which prevents the clamp from encircling the DNA substrate, they demonstrated that, depending on whether the DNA plasmid substrate threads the clamp, oxidation of the DNA takes place in a processive or distributive fashion (Fig. 26). They also demonstrate the directed loading of the clamp onto a plasmid which had been nicked using a mutated restriction enzyme. The visualisation of the DNA product was achieved using an innovative AFM approach whereby sites of oxidation were labelled with streptavidin and the product imaged directly.
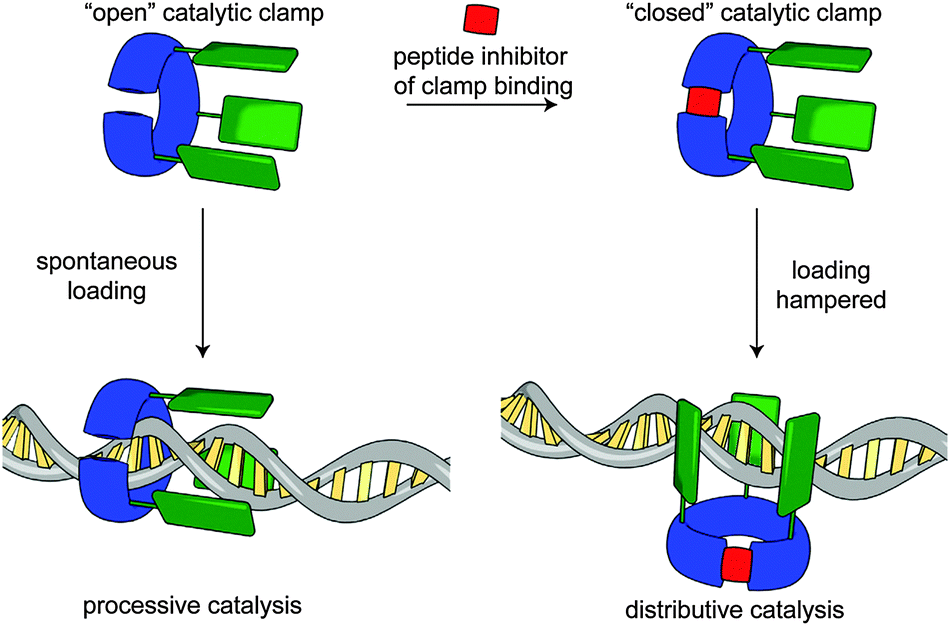 | ||
| Fig. 26 Rowan and Nolte's bio-hybrid clamp for DNA oxidation. When the clamp is prevented from encircling the DNA the oxidation events are distributed randomly along the plasmid. When threading occurs the catalyst behaves processively. From ref. 91 and reprinted with permission from Nature Publishing Group. | ||
Recently, Leigh and co-workers described a synthesising molecular machine which uses steric control over the movement of a catalyst relative to a threaded substrate to deliver the first example of a processive rotaxane molecular machine that is capable of reading a covalent template to produce a single, simple, sequence specific product (Fig. 27).92 The components of the molecular machine are the thread, which bears the amino acid monomers as reactive phenolic esters, and a macrocycle which bears both a terminal amine and a cysteine unit capable of mediating a native chemical ligation reaction. The key feature of the machine is that the amino acid monomers are too bulky to allow the macrocycle to pass without prior cleavage of the phenolic esters. Thus, the macrocycle cleaves the amino acids in the order it encounters them on the thread, to produce an activated thioester and transfers them to the amino terminus via a native chemical ligation reaction. This process repeats until all the esters have been transferred to the macrocycle and the macrocycle can escape the thread. HPLC analysis revealed the process to be remarkably selective, with only the desired tripeptide sequence observed. When the same reaction was carried out with a non-interlocked catalyst a complex mixture of products was observed which demonstrated the importance of the rotaxane architecture, which serves to inextricably link the reaction coordinate and the motion of the macrocycle.
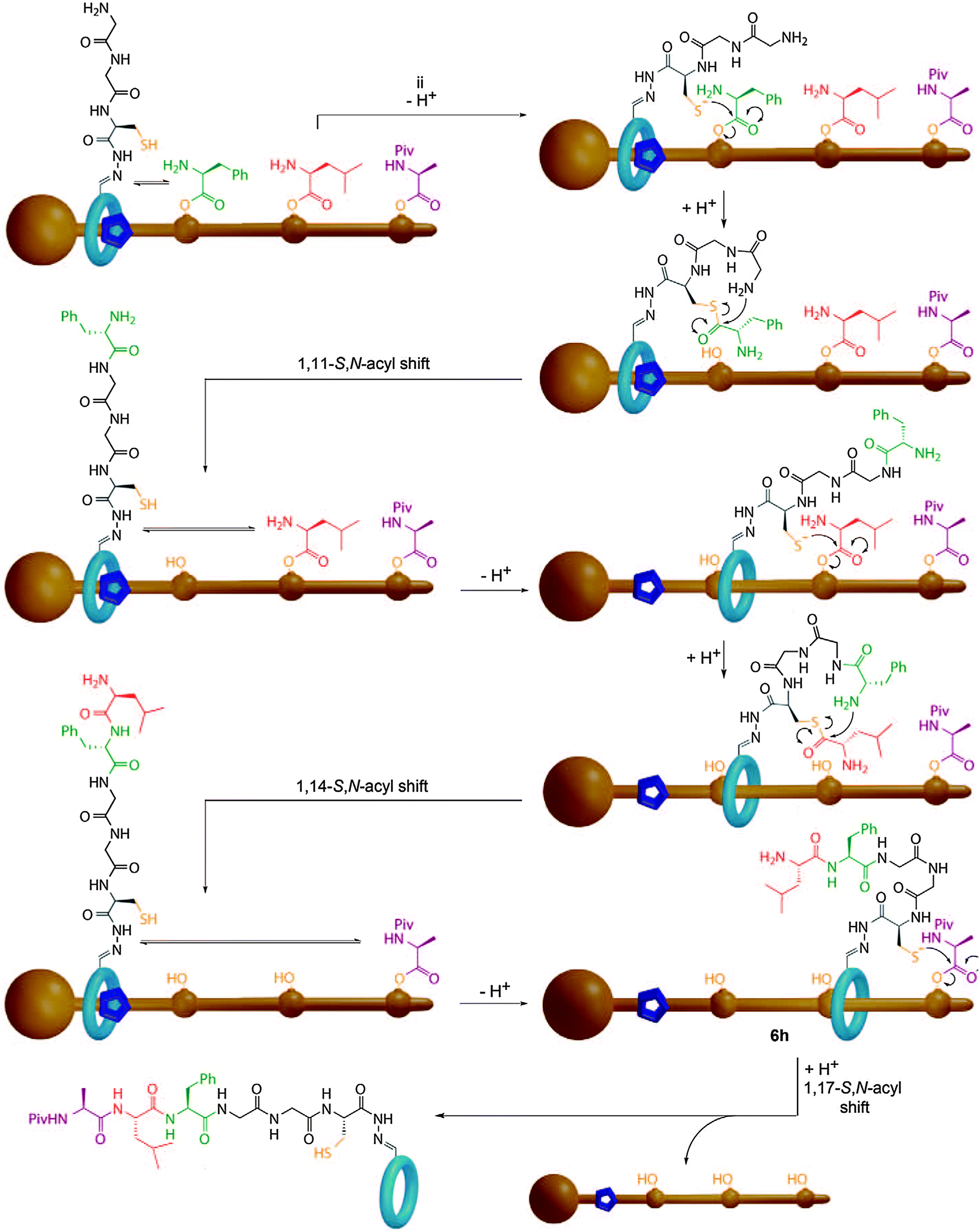 | ||
| Fig. 27 Leigh's sequence processive peptide synthesising rotaxane. The macrocycle bearing the catalyst proceeds along the thread, cleaving each amino acid ester gate in turn and transferring the activated thioester to the N-terminus of the peptide in a native chemical ligation reaction. From ref. 92 and reprinted with permission from the American Association for the Advancement of Science. | ||
8. Conclusions
In this Feature Article we have discussed some of the unusual physical and chemical properties of catenanes and rotaxanes, and highlighted some of the challenges, opportunities and applications that can result from their mechanically bonded structure. Some of these applications, such as insulated molecular wires, mechanically-stabilised dyes and slide ring polymers are well established while others, such as the use of mechanical bonding in the development of targeted pro-drugs, are already showing significant promise.Although synthetic methods for the production of mechanically bonded molecules are now mature and allow complex products, often with multiple mechanically bonded components, to be synthesised efficiently, there is still a need for methods that allow the synthesis of rotaxane and catenane stereoisomers. In particular, the synthesis of mechanically chiral molecules remains an unsolved problem which has prevented all but the most cursory investigation of their properties.
Finally, the use of rotaxanes and catenanes as catalysts for synthetic transformations is starting to receive attention and, based on these early results, is proving a rich area for investigation. Further, as demonstrated in recent work by Nolte and Rowan, and Leigh and co-workers, by combining mechanical motion with catalysis it is possible to control chemical reactions and alter their outcomes, in much the same way that nature uses controlled molecular motion to regulate complex chemical processes such as DNA transcription. These exciting results point the way to a marriage of the fields of catalysis and molecular machines, which may provide future breakthroughs for chemical synthesis.
Ultimately, artificial molecular machines that carry out multi-step chemical reactions may allow synthetic chemistry to approach the complexity of nature's synthetic machinery, which produces intricate products in an astounding mixture of starting materials and catalysts, with each performing only their desired function. In particular, molecular machines may find application in the synthesis of copolymers with a precise sequence of monomers. Such materials are produced by nature with apparent ease, but remain a challenge for conventional synthetic methodologies.
Acknowledgements
The authors thank Dr Euan Kay (St Andrews) for illuminating discussions. SMG is a Royal Society University Research Fellow.Notes and references
- E. Wasserman, J. Am. Chem. Soc., 1960, 82, 4433 CrossRef CAS.
- I. T. Harrison and S. Harrison, J. Am. Chem. Soc., 1967, 89, 5723 CrossRef CAS.
- G. Schill and A. Lüttringhaus, Angew. Chem., Int. Ed. Engl., 1964, 3, 546–547 CrossRef.
- The “[2]” refers to the number of interlocked components. Thus a [2]catenane contains two interlocked rings and a [2]rotaxane contains one axle and one macrocycle.
- C. O. Dietrich-Buchecker, J. P. Sauvage and J. P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095–5098 CrossRef CAS.
- W. R. Dichtel, O. S. Miljanić, W. Zhang, J. M. Spruell, K. Patel, I. Aprahamian, J. R. Heath and J. F. Stoddart, Acc. Chem. Res., 2008, 41, 1750–1761 CrossRef CAS PubMed; P. D. Beer, M. R. Sambrook and D. Curiel, Chem. Commun., 2006, 2105–2117 RSC; J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC; J. A. Faiz, V. Heitz and J.-P. Sauvage, Chem. Soc. Rev., 2009, 38, 422–442 RSC; A. Harada, Y. Takashima and H. Yamaguchi, Chem. Soc. Rev., 2009, 38, 875–882 RSC; K. D. Hanni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240–1251 RSC; R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203–2237 RSC; J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef PubMed; R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef PubMed; A. Coskun, J. M. Spruell, G. Barin, W. R. Dichtel, A. H. Flood, Y. Y. Botros and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 4827–4859 RSC; V. N. Vukotic and S. J. Loeb, Chem. Soc. Rev., 2012, 41, 5896–5906 RSC; G. T. Spence and P. D. Beer, Acc. Chem. Res., 2012, 46, 571–586 CrossRef PubMed.
- J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC.
- E. Wasserman and H. L. Frisch, J. Am. Chem. Soc., 1961, 83, 3789–3795 CrossRef.
- It should be noted that strictly speaking, while catenanes are topological isomers of their non-interlocked components, rotaxanes are not as the topologically allowed (although chemically forbidden) stretching of the macrocycle over the stopper units converts a rotaxane into its non-interlocked components8,31.
- A. G. Johnston, D. A. Leigh, A. Murphy, J. P. Smart and M. D. Deegan, J. Am. Chem. Soc., 1996, 7863, 10662–10663 CrossRef.
- S. Anderson and H. Anderson, Angew. Chem., Int. Ed. Engl., 1996, 35, 1956–1959 CrossRef CAS.
- M. Asakawa, G. Brancato, M. Fanti, D. A. Leigh, T. Shimizu, A. M. Z. Slawin, J. K. Y. Wong, F. Zerbetto and S. Zhang, J. Am. Chem. Soc., 2002, 124, 2939–2950 CrossRef CAS PubMed.
- It should be noted that the motion of the small ring around the large ring, commonly referred to as circumrotation, is equivalent to holding the small ring stationary and allowing the large ring to pirouette on its own axis. Thus, although differentiation of the motions of the large and small rings in hetero-[2]catenanes as circumrotation and pirouetting allows a helpful link with the shuttling and pirouetting motions in rotaxanes, in both cases the rings move relative to one another in the same way; it is just that we tend to visualise the larger component as stationary.
- C. P. Collier, G. Mattersteig, E. W. Wong, Y. Luo, K. Beverly, J. Sampaio, F. M. Raymo, J. F. Stoddart and J. R. Heath, Science, 2000, 289, 1172–1175 CrossRef CAS.
- P. Anelli, N. Spencer and J. Stoddart, J. Am. Chem. Soc., 1991, 113, 5131–5133 CrossRef CAS.
- R. Bissell, E. Córdova, A. Kaifer and J. Stoddart, Nature, 1994, 369, 133–137 CrossRef CAS.
- G. Bottari, F. Dehez, D. A. Leigh, P. J. Nash, E. M. Pérez, J. K. Y. Wong and F. Zerbetto, Angew. Chem., Int. Ed., 2003, 42, 5886–5889 CrossRef CAS PubMed.
- C. Gong and H. Gibson, Angew. Chem., Int. Ed. Engl., 1997, 2331–2333 CrossRef CAS; A. S. Lane, D. A. Leigh and A. Murphy, J. Am. Chem. Soc., 1997, 119, 11092–11093 CrossRef.
- A. Livoreil, C. O. Dietrich-Buchecker and J.-P. Sauvage, J. Am. Chem. Soc., 1994, 116, 9399–9400 CrossRef CAS; L. Raehm, J.-M. Kern and J.-P. Sauvage, Chem.–Eur. J., 1999, 5, 3310–3317 CrossRef.
- F. Vögtle, W. M. Müller, U. Müller, M. Bauer and K. Rissanen, Angew. Chem., Int. Ed. Engl., 1993, 32, 1295–1297 CrossRef.
- E. M. Pérez, D. T. F. Dryden, D. A. Leigh, G. Teobaldi and F. Zerbetto, J. Am. Chem. Soc., 2004, 126, 12210–12211 CrossRef CAS PubMed; Q.-C. Wang, D.-H. Qu, J. Ren, K. Chen and H. Tian, Angew. Chem., Int. Ed., 2004, 43, 2661–2665 CrossRef; Y. Li, H. Li, Y. Li, H. Liu, S. Wang, X. He, N. Wang and D. Zhu, Org. Lett., 2005, 7, 4835–4838 CrossRef PubMed.
- C. P. Collier, G. Mattersteig, E. W. Wong, Y. Luo, K. Beverly, J. Sampaio, F. M. Raymo, J. F. Stoddart and J. R. Heath, Science, 2000, 289, 1172–1175 CrossRef CAS.
- J. Berná, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Pérez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704–710 CrossRef PubMed.
- M. J. Chmielewski, J. J. Davis and P. D. Beer, Org. Biomol. Chem., 2009, 7, 415–424 CAS.
- R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203–2237 RSC.
- C. P. Collier, J. O. Jeppesen, Y. Luo, J. Perkins, E. W. Wong, J. R. Heath and J. F. Stoddart, J. Am. Chem. Soc., 2001, 123, 12632–12641 CrossRef CAS PubMed; A. Coskun, J. M. Spruell, G. Barin, W. R. Dichtel, A. H. Flood, Y. Y. Botros and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 4827–4859 RSC.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed; V. Balzani, A. Credi and M. Venturi, Chem. Soc. Rev., 2009, 38, 1542–1550 RSC; A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19–30 RSC.
- J. D. Crowley, D. A. Leigh, P. J. Lusby, R. T. McBurney, L.-E. Perret-Aebi, C. Petzold, A. M. Z. Slawin and M. D. Symes, J. Am. Chem. Soc., 2007, 129, 15085–15090 CrossRef CAS PubMed.
- M. N. Chatterjee, E. R. Kay and D. A. Leigh, J. Am. Chem. Soc., 2006, 128, 4058–4073 CrossRef CAS PubMed.
- J. V. Hernández, E. R. Kay and D. A. Leigh, Science, 2004, 306, 1532–1537 CrossRef PubMed.
- For a detailed discussions of chemical topology see: D. M. Walba, Tetrahedron, 1985, 41, 3161–3212 CrossRef CAS; G. A. Breault, C. A. Hunter and P. C. Mayers, Tetrahedron, 1999, 55, 5265–5293 CrossRef.
- A.-M. L. Fuller, D. A. Leigh and P. J. Lusby, J. Am. Chem. Soc., 2010, 132, 4954–4959 CrossRef CAS PubMed . For an example of the selective synthesis of one of two possible constitutional isomers of a [3]rotaxane in a single step see W. Jiang, H. D. F. Winkler and C. A. Schalley, J. Am. Chem. Soc., 2008, 130, 13852–13853 CrossRef PubMed.
- D. B. Amabilino, P. R. Ashton, A. S. Reder, N. Spencer and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1994, 33, 1286–1290 CrossRef.
- J. F. Stoddart and H. M. Colquhoun, Tetrahedron, 2008, 64, 8231–8263 CrossRef CAS PubMed.
- M. Craig, M. Hutchings, T. Claridge and H. Anderson, Angew. Chem., Int. Ed., 2001, 40, 1071–1074 CrossRef CAS; A. Arduini, F. Ciesa, M. Fragassi, A. Pochini and A. Secchi, Angew. Chem., Int. Ed., 2004, 44, 278–281 CrossRef PubMed; Q.-C. Wang, X. Ma, D.-H. Qu and H. Tian, Chem.–Eur. J., 2006, 12, 3831–3834 Search PubMed; J. W. Park, H. J. Song and H.-J. Chang, Tetrahedron Lett., 2006, 47, 3831–3834 CrossRef PubMed For an example of a [3]rotaxane stereoisomer based on two facially unsymmetrical macrocycles see: M. R. Craig, T. D. W. Claridge, H. L. Anderson and M. G. Hutchings, Chem. Commun., 1999, 1537–1538 RSC.
- G. Schill, Catenanes Rotaxanes and Knots, Academic Press, 1971 Search PubMed.
- C. Yamamoto, Y. Okamoto, T. Schmidt, R. Jager and F. Vögtle, J. Am. Chem. Soc., 1997, 7863, 10547–10548 CrossRef; D. K. Mitchell and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1988, 27, 930–931 CrossRef.
- For recent examples of the attempted selective synthesis of mechanically chiral rotaxanes see Y. Makita, N. Kihara, N. Nakakoji, T. Takata, S. Inagaki, C. Yamamoto and Y. Okamoto, Chem. Lett., 2007, 36, 162–163 CrossRef CAS; P. E. Glen, J. A. T. O'Neill and A.-L. Lee, Tetrahedron, 2013, 69, 57–68 CrossRef PubMed.
- N. Kameta, Y. Nagawa, M. Karikomi and K. Hiratani, Chem. Commun., 2006, 3714–3716 RSC.
- C. P. McArdle, S. Van, M. C. Jennings and R. J. Puddephatt, J. Am. Chem. Soc., 2002, 124, 3959–3965 CrossRef CAS PubMed.
- A. Theil, C. Mauve, M.-T. Adeline, A. Marinetti and J.-P. Sauvage, Angew. Chem., Int. Ed., 2006, 45, 2104–2107 CrossRef CAS PubMed.
- G. Agam and A. Zilkha, J. Am. Chem. Soc., 1976, 256, 5214–5216 CrossRef.
- S. J. Rowan and J. F. Stoddart, J. Am. Chem. Soc., 1999, 122, 164–165 CrossRef.
- S. J. Rowan, S. J. Cantrill, J. F. Stoddart, A. J. P. White and D. J. Williams, Org. Lett., 2000, 2, 759–762 CrossRef CAS PubMed.
- N. Kihara, S. Motoda, T. Yokozawa and T. Takata, Org. Lett., 2005, 7, 1199–1202 CrossRef CAS PubMed.
- J. S. Hannam, S. M. Lacy, D. A. Leigh, C. G. Saiz, A. M. Z. Slawin and S. G. Stitchell, Angew. Chem., Int. Ed., 2004, 43, 3260–3264 CrossRef CAS PubMed.
- D. W. Zehnder II and D. B. Smithrud, Org. Lett., 2001, 16, 2485–2487 CrossRef PubMed.
- A. Altieri, V. Aucagne, R. Carrillo, G. J. Clarkson, D. M. D'Souza, J. A. Dunnett, D. A. Leigh and K. M. Mullen, Chem. Sci., 2011, 2, 1922–1928 RSC.
- S.-Y. Hsueh, J.-L. Ko, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Angew. Chem., Int. Ed., 2011, 50, 6643–6646 CrossRef CAS PubMed.
- For an example of shrinking of a macrocycle by coordination of a transition metal ion see I. Yoon, M. Narita, T. Shimizu and M. Asakawa, J. Am. Chem. Soc., 2004, 126, 16740–16741 CrossRef CAS PubMed.
- N. Watanabe, N. Kihara and T. Takata, Org. Lett., 2001, 3, 3519–3522 CrossRef CAS PubMed.
- P.-F. Cao, A. Bunha, J. Mangadlao, M. J. Felipe, K. I. Mongcopa and R. Advincula, Chem. Commun., 2012, 48, 12094–12096 RSC.
- A. M. Albrecht-Gary, Z. Saad, C. O. Dietrich-Buchecker and J. P. Sauvage, J. Am. Chem. Soc., 1985, 107, 3205–3209 CrossRef CAS.
- Y. Tachibana, H. Kawasaki, N. Kihara and T. Takata, J. Org. Chem., 2006, 71, 5093–5104 CrossRef CAS PubMed.
- H. Lahlali, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2011, 50, 4151–4155 CrossRef CAS PubMed.
- A. H. Parham, B. Windisch and F. Vögtle, Eur. J. Org. Chem., 1999, 1233–1238 CrossRef CAS.
- D. A. Leigh and E. M. Perez, Chem. Commun., 2004, 2262–2263 RSC.
- A. Mateo-Alonso, P. Brough and M. Prato, Chem. Commun., 2007, 1412–1414 RSC.
- T. Oku, Y. Furusho and T. Takata, Org. Lett., 2003, 5, 12384–12385 CrossRef PubMed.
- J. Winn, A. Pinczewska and S. M. Goldup, J. Am. Chem. Soc., 2013, 135, 13318–13321 CrossRef CAS PubMed.
- D. M. D'Souza, D. A. Leigh, L. Mottier, K. M. Mullen, F. Paolucci, S. J. Teat and S. Zhang, J. Am. Chem. Soc., 2010, 132, 9465–9470 CrossRef CAS PubMed.
- S. Anderson, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed. Engl., 1997, 36, 1310–1313 CrossRef CAS; J. E. H. Buston, J. R. Young and H. L. Anderson, Chem. Commun., 2000, 905–906 RSC.
- E. Arunkumar, C. C. Forbes, B. C. Noll and B. D. Smith, J. Am. Chem. Soc., 2005, 127, 3288–3289 CrossRef CAS PubMed; J. J. Gassensmith, J. M. Baumes and B. D. Smith, Chem. Commun., 2009, 6329–6338 RSC.
- F. Cacialli, J. S. Wilson, J. J. Michels, C. Daniel, C. Silva, R. H. Friend, N. Severin, P. Samorì, J. P. Rabe, M. J. O'Connell, P. N. Taylor and H. L. Anderson, Nat. Mater., 2002, 1, 160–164 CrossRef CAS PubMed.
- M. J. Frampton and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 1028–1064 CrossRef CAS PubMed.
- R. Eelkema, K. Maeda, B. Odell and H. L. Anderson, J. Am. Chem. Soc., 2007, 129, 12384–12385 CrossRef CAS PubMed.
- J. C. Barnes, A. C. Fahrenbach, D. Cao, S. M. Dyar, M. Frasconi, M. A. Giesener, D. Benítez, E. Tkatchouk, O. Chernyashevskyy, W. H. Shin, H. Li, S. Sampath, C. L. Stern, A. A. Sarjeant, K. J. Hartlieb, Z. Liu, R. Carmieli, Y. Y. Botros, J. W. Choi, A. M. Z. Slawin, J. B. Ketterson, M. R. Wasielewski, W. A. Goddard III and J. F. Stoddart, Science, 2013, 339, 429–433 CrossRef CAS PubMed.
- H. Li, Z. Zhu, A. C. Fahrenbach, B. M. Savoie, C. Ke, J. C. Barnes, J. Lei, Y.-L. Zhao, L. M. Lilley, T. J. Marks, M. A. Ratner and J. F. Stoddart, J. Am. Chem. Soc., 2012, 135, 456–467 CrossRef PubMed.
- A. Fernandes, A. Viterisi, F. Coutrot, S. Potok, D. A. Leigh, V. Aucagne and S. Papot, Angew. Chem., Int. Ed., 2009, 121, 6565–6569 CrossRef.
- For a previous study on the cleavage of stopper groups from a [2]rotaxane using enzymatic cleavage and the effect of shuttling on the reaction rate see A. G. Cheetham, M. G. Hutchings, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed., 2006, 45, 1596–1599 CrossRef CAS PubMed.
- A. Fernandes, A. Viterisi, V. Aucagne, D. A. Leigh and S. Papot, Chem. Commun., 2012, 48, 2083–2085 RSC.
- K. Ito, Polym. J., 2007, 39, 489–499 CrossRef CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 8656, 485–487 CrossRef.
- J. M. Baumes, J. J. Gassensmith, J. Giblin, J.-J. Lee, A. G. White, W. J. Culligan, W. M. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025–1030 CrossRef CAS PubMed.
- J. M. Baumes, I. Murgu, A. Oliver and B. D. Smith, Org. Lett., 2010, 12, 4980–4983 CrossRef CAS PubMed.
- For an example of photochemically driven information ratchet see V. Serreli, C.-F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523–527 CrossRef CAS PubMed.
- M. Yamazaki, T. Hagiwara, M. Sekiguchi, T. Sawaguchi and S. Yano, Synth. Commun., 2008, 38, 553–558 CrossRef CAS.
- G. Hattori, T. Hori, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2007, 129, 12930–12931 CrossRef CAS PubMed.
- Y. Li, Y. Feng, Y.-M. He, F. Chen, J. Pan and Q.-H. Fan, Tetrahedron Lett., 2008, 49, 2878–2881 CrossRef CAS PubMed.
- It is noteworthy that pseudorotaxane 35 possesses both covalent axial chirality and mechanical planar chirality due to the rotationally unsymmetrical nature of the macrocycle combined with the translationally unsymmetrical thread (vide supra). The authors report that pseudo rotaxane 35 is formed as a single diastereoisomer, although which isomer was not determined. This implies that the covalent chiral information in the macrocycle has served to arrange the thread and macrocycle in a single orientation. This may explain the enhanced stereoselectivity observed in the hydrogenation reaction.
- C. B. Caputo, K. Zhu, V. N. Vukotic, S. J. Loeb and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 960–963 CrossRef CAS PubMed.
- J. Berná, M. Alajarín and R.-A. Orenes, J. Am. Chem. Soc., 2010, 132, 10741–10747 CrossRef PubMed.
- Y. Suzaki, K. Shimada, E. Chihara, T. Saito, Y. Tsuchido and K. Osakada, Org. Lett., 2011, 13, 3774–3777 CrossRef CAS PubMed.
- V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, Angew. Chem., Int. Ed., 2012, 51, 5166–5169 CrossRef CAS PubMed.
- W. A. Breyer and B. W. Matthews, Protein Sci., 2001, 10, 1699–1711 CrossRef CAS PubMed.
- H. Makis and A. Kornberg, J. Biol. Chem., 1985, 260, 12987–12992 Search PubMed.
- P. T. Stukenberg, P. S. Studwell-Vaughan and M. O'Donnell, J. Biol. Chem., 1991, 266, 11328–11334 CAS.
- P. Thordarson, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Nature, 2003, 424, 915–918 CrossRef CAS PubMed; P. H. Ramos, R. G. E. Coumans, A. B. C. Deutman, J. M. M. Smits, R. de Gelder, J. A. A. W. Elemans, R. J. M. Nolte and A. E. Rowan, J. Am. Chem. Soc., 2007, 129, 5699–5702 CrossRef PubMed.
- R. G. E. Coumans, J. A. A. W. Elemans, R. J. M. Nolte and A. E. Rowan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19647–19651 CrossRef CAS PubMed.
- N. Miyagawa, M. Watanabe, T. Matsuyama, Y. Koyama, T. Moriuchi, T. Hirao, Y. Furusho and T. Takata, Chem. Commun., 2010, 46, 1920–1922 RSC.
- S. F. M. van Dongen, J. Clerx, K. Nørgaard, T. G. Bloemberg, J. J. L. M. Cornelissen, M. a. Trakselis, S. W. Nelson, S. J. Benkovic, A. E. Rowan and R. J. M. Nolte, Nat. Chem., 2013, 1–8 Search PubMed.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |