 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvidence that steric factors modulate reactivity of tautomeric iron–oxo species in stereospecific alkane C–H hydroxylation†
Mainak
Mitra
a,
Julio
Lloret-Fillol
b,
Matti
Haukka
c,
Miquel
Costas
*b and
Ebbe
Nordlander
*a
aChemical Physics, Department of Chemistry, Lund University, P.O. Box 124, Lund, SE-221 00, Sweden. E-mail: Ebbe.Nordlander@chemphys.lu.se; Fax: +46-46-22-24119; Tel: +46-46-22-28118
bQBIS Group, Department of Chemistry and Institut de Quimica Computacional i Catalisi (IQCC), Universitat de Girona, Campus Montilivi, 17071 Girona, Catalonia, Spain. E-mail: miquel.costas@udg.edu; Fax: +34-972-41-81-50; Tel: +34-972-41-98-42
cUniversity of Jyväskylä, Department of Chemistry, P.O. Box 35, FI-40014 University of Jyväskylä, Finland
First published on 4th November 2013
Abstract
A new iron complex mediates stereospecific hydroxylation of alkyl C–H bonds with hydrogen peroxide, exhibiting excellent efficiency. Isotope labelling studies provide evidence that the relative reactivity of tautomerically related oxo–iron species responsible for the C–H hydroxylation reaction is dominated by steric factors.
While the selective functionalization of hydrocarbons remains a significant challenge for chemists,1 several iron-dependent oxygenases are able to mediate the hydroxylation of C–H bonds under mild conditions, using dioxygen as the terminal oxidant.2 Examples include the cytochrome P450 enzymes,3 and the family of non-heme iron-dependent Rieske oxygenases.4 In both cases, C–H hydroxylation occurs with almost complete stereoretention, and is accomplished via the intermediacy of an electrophilic high valent iron–oxo species that attacks the C–H bond via the so-called oxygen-rebound mechanism (Scheme 1).5
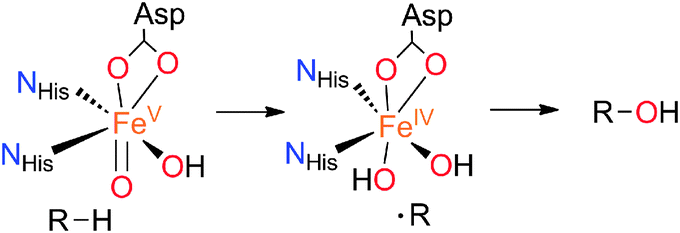 | ||
| Scheme 1 Schematic mechanism for C–H hydroxylation by a Rieske oxygenase enzyme. | ||
A fundamental difference between heme and non-heme sites is that active sites in the latter contain lower coordination numbers, and a number of them form reactive intermediates containing a cis-Fe(O)(X) unit (X = HO(H), Cl, Br). This leads to a versatile reactivity that opens new mechanistic scenarios. Arene cis-dihydroxylation and aliphatic chlorination constitute unique examples of the reactivity exhibited by cis-Fe(O)(X) units (X = OH, Cl and Br).4,5b,6
The reactivity of non-heme oxygenases has inspired the design of synthetic model complexes as selective C–H oxidation catalysts.7 Mechanistic studies have shown that in selected cases reactions are metal based, involving high-valent oxo–iron species, and are fundamentally distinct from radical pathway Fenton processes.8 The Fe(PyTACN) family of complexes (Scheme 2) belongs to the group of catalysts that mediate C–H hydroxylation with retention of configuration.8d We and others have proposed a mechanistic scenario resembling the “peroxide shunt”3 of cytochrome P450 and model systems. A highly electrophilic [FeV(O)(OH)(LN4)]2+ oxidant (O), formed via water-assisted cleavage of a hydroperoxide [FeIII(OOH)(OH2)(LN4)]2+ (PB) (Scheme 2), is ultimately responsible for C–H oxidation reactions.8a,d,9–11 Intermediate O can exist as two tautomerically related species, OA and OB, that differ in the relative positions of the oxo and hydroxide ligands, and are connected through prototopic oxo–hydroxo tautomerism. We have also previously studied C–H oxidation reactions using a set of catalysts where electronic properties of the PyTACN ligand were systematically tuned, and found that the relative reactivity of OA and OB in C–H oxidations remains basically the same, irrespective of the catalyst.8d
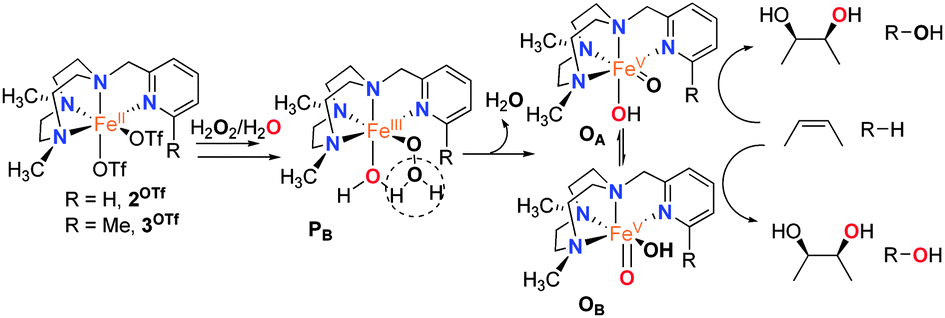 | ||
| Scheme 2 Mechanism for substrate oxidation by Fe(PyTACN) complexes. | ||
In this work we turn our attention towards investigation of steric effects. Towards this end, C–H oxidation reactions catalyzed using the new iron complex [FeII(CF3SO3)2(Me2,BzImTACN)] (Fig. 1), 1OTf, were studied. The new tetradentate ligand Me2,BzImTACN has been developed by replacing the pyridyl arm of the PyTACN scaffold by an N-methyl benzimidazolyl substituent. The sp2 character and the rigidity of the latter substituent should provide a well-defined steric demand, intermediate between the α-H and the α-Me groups of a pyridine (Scheme 2, catalysts 2OTf and 3OTf). On the other hand, the relative donor capacities of pyridine and benzimidazole can be estimated to be very similar by comparing the pKa values of their conjugate acids (5.22 for pyridine, 5.41 for benzimidazole and 5.57 for α-Me pyridine), and therefore differences in reactivity among this set of complexes can be traced to steric factors.
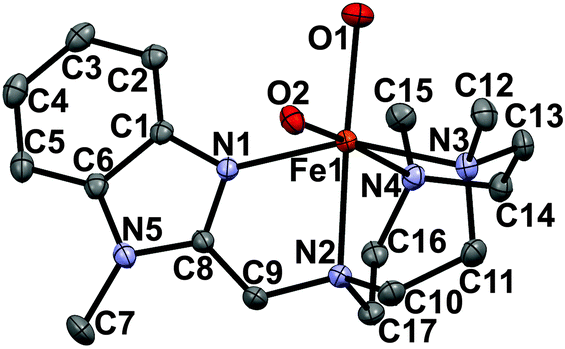 | ||
| Fig. 1 Molecular structure of [FeII(H2O)2(Me2,BzImTACN)]2+ (1H2O) with 30% probability ellipsoids; H-atoms have been omitted for clarity. | ||
The complexes [FeII(CF3SO3)2(Me2,BzImTACN)], 1OTf, and [FeII(H2O)2(Me2,BzImTACN)](CF3SO3)2, 1H2O, were prepared and characterized following standard procedures (see ESI† for details). The X-ray structures of 1OTf and 1H2O are very similar to those of 2OTf and 3OTf and have an iron center in a distorted octahedral environment surrounded by the four N atoms of the ligand, with the TACN ring capping one face of the octahedron, and two oxygen atoms of triflate anions (1OTf) or water molecules (1H2O) cis to each other (cf.Fig. 1 and ESI†).8d Average Fe–NTACN and Fe–OH2O distances are 2.23 Å and 2.13 Å respectively, characteristic of a high spin ferrous center.12
Complex 1OTf was found to be an outstanding catalyst in C–H oxidation reactions with H2O2. Catalytic oxidation of cyclohexane was chosen for appropriate comparison with literature precedents.8,13 Syringe pump addition of 10 equivalents (w.r.t. the complex) of H2O2 together with 1000 equivalents of H2O‡ to a CH3CN solution containing 1 and a substrate (1000 equivalents) over 30 min in air at room temperature resulted in the formation of cyclohexanol (A) with a turnover number (TON) of 8.5 and a small amount of cyclohexanone (K) with a TON of 0.8, giving an alcohol/ketone (A/K) ratio of 10.6. The efficiency w.r.t. consumption of the oxidant was around 99–100%, and remains unusually high (54%) when 100 equiv. of H2O2 are employed. Interestingly, when followed over time, the A/K product ratio in oxidation of cyclohexane showed that the initial value of A/K was around 35, which gradually decreased to 10.6 (cf. Fig. S5, ESI†). This provides strong evidence that cyclohexanol is the almost exclusive primary product of the alkane oxidation reaction, and cyclohexanone is a result of further oxidation of the alcohol, thereby eliminating the significant implication of a Russell-type termination mechanism initiated by hydroxyl radicals and producing equal amounts of alcohol and ketone.
Several mechanistic probes further substantiate that the reactions are metal-based. The intermolecular kinetic isotope effect was determined for the formation of cyclohexanol from a mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) of cyclohexane and its deuterated isotopomer cyclohexane-d12, and was found to be 5. Also, complex 1OTf oxidizes adamantane with a large C3/C2 normalized selectivity (14) towards the tertiary C–H bond. The oxidation of cis-1,2-dimethylcyclohexane (DMCH) leads to the corresponding tertiary alcohol product with 97% retention of configuration. These data are consistent with the implication of selective oxidants whose relative reactivities against C–H bonds are modulated by their bond strengths and steric properties.7a The reactivity of 1OTf against these mechanistic probes is thus in good accordance with that described for iron catalysts that mediate stereospecific C–H hydroxylation, including those of the [Fe(PyTACN)] family.8d Since these catalysts operate via a [FeV(O)(OH)(LN4)]2+ (O) oxidant,8a,d,10,11 the same was tentatively inferred for 1OTf. Strong experimental evidence in favor of this scenario arises from olefin cis-dihydroxylation reactions. The water assisted cleavage of the O–O bond (Scheme 2) determines the oxygen atom inventory in the HO–FeV
:3) of cyclohexane and its deuterated isotopomer cyclohexane-d12, and was found to be 5. Also, complex 1OTf oxidizes adamantane with a large C3/C2 normalized selectivity (14) towards the tertiary C–H bond. The oxidation of cis-1,2-dimethylcyclohexane (DMCH) leads to the corresponding tertiary alcohol product with 97% retention of configuration. These data are consistent with the implication of selective oxidants whose relative reactivities against C–H bonds are modulated by their bond strengths and steric properties.7a The reactivity of 1OTf against these mechanistic probes is thus in good accordance with that described for iron catalysts that mediate stereospecific C–H hydroxylation, including those of the [Fe(PyTACN)] family.8d Since these catalysts operate via a [FeV(O)(OH)(LN4)]2+ (O) oxidant,8a,d,10,11 the same was tentatively inferred for 1OTf. Strong experimental evidence in favor of this scenario arises from olefin cis-dihydroxylation reactions. The water assisted cleavage of the O–O bond (Scheme 2) determines the oxygen atom inventory in the HO–FeV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxidant (O). One of the oxygen atoms originates from the water molecule, while the second oxygen atom is derived from the peroxide. cis-Dihydroxylation reactions incorporate both oxygen atoms of O into the product and consequently syn-diols must contain one oxygen atom that originates from water and one oxygen from the peroxide.11 Indeed, 1OTf catalyzes the oxidation of cyclooctene (1OTf:H2O2:H218O:cyclooctene, 1:10:1000:1000) affording cis-cyclooctene epoxide (TON = 2) and syn-cyclooctane-1,2-diol (TON = 7). The syn-diol is 98% 16O18O labeled, providing strong support in favor of O as the oxidant.
O oxidant (O). One of the oxygen atoms originates from the water molecule, while the second oxygen atom is derived from the peroxide. cis-Dihydroxylation reactions incorporate both oxygen atoms of O into the product and consequently syn-diols must contain one oxygen atom that originates from water and one oxygen from the peroxide.11 Indeed, 1OTf catalyzes the oxidation of cyclooctene (1OTf:H2O2:H218O:cyclooctene, 1:10:1000:1000) affording cis-cyclooctene epoxide (TON = 2) and syn-cyclooctane-1,2-diol (TON = 7). The syn-diol is 98% 16O18O labeled, providing strong support in favor of O as the oxidant.
Having obtained positive evidence that 1OTf operates through the same mechanism as that of 2OTf and 3OTf, we proceeded to investigate the relative reactivity of the OA/OB tautomers in C–H hydroxylation reactions. Since the origin of the oxygen atoms is determined in the peroxide precursor (PB), the relative reactivity of OA and OB in C–H hydroxylation can be probed using isotopically labeled water and hydrogen peroxide (Scheme 2). The oxidation of cyclohexane by 1OTf in the presence of 10 equivalents of H218O2 and 1000 equivalents of H2O afforded 45% 18O-labeled cyclohexanol. Complementary experiments with 10 equivalents of H2O2 and 1000 equivalents of H218O afforded 48% 18O-labeled cyclohexanol (Table 1).
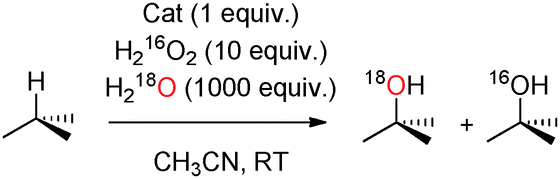
Similar levels of 18O-label incorporation from H218O were observed in the case of cyclooctane (41%) and cyclohexane-d12 (48%). These levels of water incorporation are unusually high, only bypassed in the literature by 2OTf,8d and constitute strong evidence that OA and OB are roughly equally reactive against secondary C–H bonds. Most interestingly, when the substrates contain tertiary C–H bonds (e.g. DMCH and adamantane), the percentages of 18O incorporation from H218O were found to be in the range 25–29%, indicating a preferential oxidation viaOA.
Interpretation of these values can be done by considering those obtained using 2OTf and 3OTf in analogous reactions. Table 1 shows that hydroxylation of tertiary C–H bonds mediated by 2OTf is predominantly performed by OB as shown by the large extent of oxygen atoms originating from water in the alcohol product (up to 79%, cf.Table 1). Instead, hydroxylation of secondary C–H bonds occurs with incorporation of ∼40% of oxygen atoms from water, suggesting comparable reactivity of both tautomers. In sharp contrast, hydroxylation catalyzed by 3OTf exhibits a relatively small extent (∼10%) of water incorporation in hydroxylation of secondary C–H bonds, and negligible (<3%) incorporation in the hydroxylation of tertiary C–H sites, indicating that hydroxylation is almost exclusively performed by OA.
Therefore, the relative reactivity of the two tautomeric forms of the [FeV(O)(OH)(LN4)]2+ (O) intermediate is finely tuned among the series of catalysts (1OTf–3OTf), a fact that contrasts with the small effects exerted when the electronic properties of the pyridine in a series of catalysts is altered.8d Trends observed for 1OTf–3OTf may thus be rationalized on the basis of steric effects. The benzylimidazole ring introduces steric bulk in the proximity of position B at the iron center that is intermediate between that set by pyridine and 6-Me-pyridine arms (Fig. 2). Accordingly, when secondary C–H bonds are hydroxylated, 1OTf behaves as 2OTf, i.e. tautomers OA and OB are equally implicated in the C–H oxidation reaction. On the other hand, oxidation of sterically more demanding tertiary C–H bonds yields levels of water incorporation that suggest predominant participation of OA as in the case of 3OTf, although unlike in the latter case, implication of OB remains significant (∼25%) because steric hindrance at position B induced by the C–C-sp2 benzylimidazole moiety is smaller than the one caused by a C-sp3 methyl substituent.
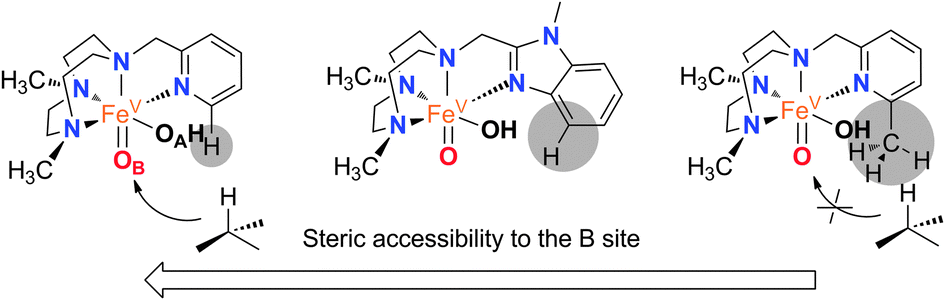 | ||
| Fig. 2 Comparative analysis of the steric bulk in proximity to site B. | ||
In conclusion, the present work adds to the growing evidence that the coordination environment at non-heme sites opens reactivity scenarios unattainable by hemes. Here we have shown that systematic tuning of the steric properties of the two sites in the cis-Fe(O)(X) unit translates into systematic differences in relative reactivity of the two iron–oxo tautomers. We postulate that analogous steric conditions may influence the relative reactivities of putative tautomers in non-heme iron oxygenases.
This work has been supported by the European Union (the Erasmus Mundus program), the International Research Training Group Metal Sites in Biomolecules: Structures, Regulation and Mechanisms (www.biometals.eu), and COST Action CM1003. M.C. acknowledges ERC-29910, MINECO of Spain for CTQ2012-37420-C02-01/BQU and CSD2010-00065, catalan DIUE (2009SGR637) and an ICREA academia award. J.Ll. thanks MICINN for a RyC contract. We thank Prof. Albert A. Shteinman for fruitful discussions and Dr Santanu Mandal for 13C-NMR measurements.
Notes and references
- (a) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed; (b) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (c) M. Bordeaux, A. Galarneau and J. Drone, Angew. Chem., Int. Ed., 2012, 51, 10712–10723 CrossRef CAS PubMed.
- (a) C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS PubMed; (b) M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr., Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed; (c) M. M. Abu-Omar, A. Loaiza and N. Hontzeas, Chem. Rev., 2005, 105, 2227–2252 CrossRef CAS PubMed; (d) E. G. Kovaleva and J. D. Lipscomb, Nat. Chem. Biol., 2008, 4, 186–193 CrossRef CAS PubMed.
- (a) J. T. Groves, Cytochrome P-450. Structure, Mechanism, and Biochemistry, ed. P. R. Ortiz de Montellano, Plenum Press, New York, 2005, pp. 1–43 Search PubMed; (b) P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed.
- (a) D. T. Gibson and R. E. Parales, Curr. Opin. Biotechnol., 2000, 11, 236–243 CrossRef CAS; (b) A. Karlsson, J. V. Parales, R. E. Parales, D. T. Gibson, H. Eklund and S. Ramaswamy, Science, 2003, 299, 1039–1042 CrossRef CAS PubMed.
- (a) J. T. Groves, G. A. McClusky, R. E. White and M. J. Coon, Biochem. Biophys. Res. Commun., 1978, 81, 154–160 CrossRef CAS; (b) S. Chakrabarty, R. N. Austin, D. Deng, J. T. Groves and J. D. Lipscomb, J. Am. Chem. Soc., 2007, 129, 3514–3515 CrossRef CAS PubMed.
- D. P. Galonić, E. W. Barr, C. T. Walsh, J. M. Bollinger and C. Krebs, Nat. Chem. Biol., 2007, 3, 113–116 CrossRef PubMed.
- (a) L. Que and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef CAS PubMed; (b) M. C. White, Science, 2012, 335, 807–809 CrossRef CAS PubMed; (c) E. B. Bauer, Curr. Org. Chem., 2008, 12, 1341–1369 CrossRef CAS.
- (a) K. Chen and L. Que Jr., J. Am. Chem. Soc., 2001, 123, 6327–6337 CrossRef CAS PubMed; (b) J. Yoon, S. A. Wilson, Y. K. Jang, M. S. Seo, K. Nehru, B. Hedman, K. O. Hodgson, E. Bill, E. I. Solomon and W. Nam, Angew. Chem., Int. Ed., 2009, 48, 1257–1260 CrossRef CAS PubMed; (c) S. H. Lee, J. H. Han, H. Kwak, S. J. Lee, E. Y. Lee, H. J. Kim, J. H. Lee, C. Bae, S. N. Lee, Y. Kim and C. Kim, Chem.–Eur. J., 2007, 13, 9393–9398 CrossRef CAS PubMed; (d) I. Prat, A. Company, V. Postils, X. Ribas, L. Que, J. M. Luis and M. Costas, Chem.–Eur. J., 2013, 19, 6724–6738 CrossRef CAS PubMed; (e) Y. Hitomi, K. Arakawa, T. Funabiki and M. Kodera, Angew. Chem., Int. Ed., 2012, 51, 3448–3452 CrossRef CAS PubMed.
- DFT calculations indicate that the isomeric peroxide PA is not implicated in chemistry. See ref. 8d.
- (a) D. Quinonero, K. Morokuma, D. G. Musaev, R. Mas-Balleste and L. Que Jr., J. Am. Chem. Soc., 2005, 127, 6548–6549 CrossRef CAS PubMed; (b) A. Bassan, M. R. A. Blomberg, P. E. M. Siegbahn and L. Que Jr., J. Am. Chem. Soc., 2002, 124, 11056–11063 CrossRef CAS PubMed.
- (a) I. Prat, J. S. Mathieson, M. Guell, X. Rivas, J. M. Luis, L. Cronin and M. Costas, Nat. Chem., 2011, 3, 788–793 CrossRef CAS PubMed; (b) W. N. Oloo, A. J. Fielding and L. Que Jr., J. Am. Chem. Soc., 2013, 135, 6438–6441 CrossRef CAS PubMed.
- (a) D. Blakesley, S. C. Payne and K. S. Hagen, Inorg. Chem., 2000, 39, 197–198 CrossRef; (b) Y. Zang, J. Kim, Y. Dong, E. C. Wilkinson, E. H. Appelman and L. Que Jr., J. Am. Chem. Soc., 1997, 119, 4197–4205 CrossRef CAS.
- (a) J. England, C. R. Davies, M. Banaru, A. J. P. White and G. J. P. Britovsek, Adv. Synth. Catal., 2008, 350, 883–897 CrossRef CAS; (b) G. J. P. Britovsek, J. England and A. J. P. White, Inorg. Chem., 2005, 44, 8125–8134 CrossRef CAS PubMed; (c) P. Comba, M. Maurer and P. Vadivelu, Inorg. Chem., 2009, 48, 10389–10396 CrossRef CAS PubMed; (d) Y. Mekmouche, S. Ménage, C. Toia-Duboc, M. Fontecave, J.-B. Galey, C. Lebrun and J. Pecaut, Angew. Chem., Int. Ed., 2001, 40, 949–952 CrossRef CAS; (e) Y. He, J. D. Gorden and C. R. Goldsmith, Inorg. Chem., 2011, 50, 12651–12660 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Ligand synthesis, complex synthesis, proton NMR spectra, ESI-MS and IR spectra of the complex, crystallographic data for complexes 1OTf and 1H2O, catalysis experiments and results and details of isotope labelling experiments. CCDC 960138 and 960139. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc47830k |
| ‡ Analogous product yields and A/K selectivity values were obtained when H2O (1000 equiv.) was not specifically added. |
| This journal is © The Royal Society of Chemistry 2014 |