 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unprecedented participation of a four-coordinate hydrogen atom in the cubane core of lithium and sodium phenolates†
David M.
Cousins
,
Matthew G.
Davidson
* and
Daniel
García-Vivó
*
Centre for Sustainable Chemical Technologies, Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: m.g.davidson@bath.ac.uk; garciavdaniel@uniovi.es
First published on 24th October 2013
Abstract
Metalation of an ammonium tris(phenol) ligand affords new lithium/sodium derivatives having central pseudocubane motifs in which one of the metallic positions is replaced by a four-coordinate hydrogen atom.
Periodic trends in the chemical properties of elements are one of the most important and better established concepts in chemistry.1 They are elegantly represented in the Periodic table, which provides a visual representation of the connection between atomic number, electronic structure and group relationships. One of the best examples of group trends is found for the alkali metals which exhibit well-defined changes in their atomic properties.1 However, the position of hydrogen as the first element of Group 1 is a question which is still open to debate. Undoubtedly, the similarity of the electronic structure of H (1s1) with that of the alkaline elements (ns1) is not, at least obviously, translated into a similarity in the properties of these elements (i.e. under ambient conditions the former is a diatomic gas and the latter are solid metals). In spite of this, over the years a close relationship between hydrogen and the light elements in the group (mainly Li) has been discerned in two respects. First, under extreme conditions, hydrogen can exhibit “metallic” properties typical of the alkali metals2 and, secondly, lithium has been attributed H-like characteristics. Thus, while hydrogen has been known for decades to participate in secondary interactions known as “hydrogen bonds” the analogous “lithium bond” was first suggested in 19583 and predicted theoretically by Kollman et al. in 1970.4 Since then the analogy has been generally accepted and “lithium bonds” have been the basis of many theoretical and spectroscopic investigations.5 These similarities are not only important for a better understanding of fundamental chemical principles, but have also recently received attention in organocatalysis, after considering that hydrogen atoms can perform the role of Lewis acidic metal centres.6
Given these similarities, as well as recent advances in synthetic methodology, and the plethora of organic and metal–organic crystal structure data available, an obvious question to ask is why have hydrogen and the alkali metals rarely, if ever, been observed to perform the same structural role in a molecular crystal structure?
One answer to this question lies in the difference in size which leads to very different structural preferences (Chart 1). Thus, the smaller H atom, when covalently bonded to an electronegative atom, has a strong preference for a nearly linear two-centre secondary interaction with another electronegative centre. Higher coordination numbers are possible via additional secondary interactions, although the occurrence of three-centre hydrogen bonds (bifurcated) is less common and four-centre (trifurcated) hydrogen bonds are rarely observed in organic crystals (Chart 1).7 By comparison, heavier Group 1 elements are larger atoms which can easily accommodate higher coordination numbers and they almost exclusively participate in three and four-centre interactions via either association (oligomers), coordination of neutral donor ligands (solvation) or commonly by a combination of both (Chart 1).8 As a result, one of the most common structural motifs for these elements is the solvated pseudocubane.
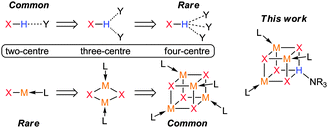 | ||
| Chart 1 Typical structural motifs for hydrogen (top) and Li/Na (bottom). | ||
In order to induce a structural equivalence of H and the alkali metals, one approach might be using a ligand framework combining the preference for association into the pseudocubane structural motif with the ability to force H into a trifurcated bonding mode. Overcoming the fundamental structural differences in this way would provide the opportunity for H to participate in a molecular metallic array typical of the alkali metals (behaving as an alkali metal structurally). As part of our studies of metal complexes of triphenolamines (Scheme 1),9 here we report the synthesis and characterisation of multi-lithiated and multi-sodiated derivatives of these ligands which display some unique structural features. This includes the first examples of complexes in which a H atom participates in the pseudocubane motif typical of Li/Na aryloxides8,10 by replacing one of the metallic positions, proving for the first time that a structural equivalence between these elements is indeed possible.
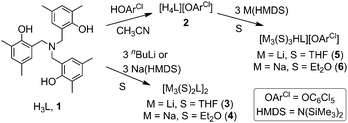 | ||
| Scheme 1 Synthesis of new compounds. | ||
The neutral tris(phenol) ligand 1 (Scheme 1) is readily prepared in high yield following protocols provided in the literature.11 Addition of one equivalent of an acidic enough phenol, such as pentachlorophenol (HOArCl), to acetonitrile solutions of 1 immediately induces the precipitation of the corresponding ammonium phenolate ligand, [H4L]OArCl (2), as a white solid in quantitative yield. Ligand 2 has been characterized by comparing its NMR data with that available for the crystallographically characterized chloride salt [H4L]Cl.12 A common feature of these two salts is the appearance of highly deshielded resonances in their 1H NMR spectra [9.28 (Cl) vs. 9.32 (2) ppm], corresponding to the N–H group, a feature which remains unaltered in the metallated derivatives of 2 as discussed later on.
We then turned our attention to the synthesis of alkali metal derivatives of these two ligands. Although the tri(lithium) and tri(sodium) derivatives of 1 have recently been prepared and used (in situ) as starting materials for the preparation of a variety of transition-metal derivatives of 1,13 no structural characterization has been reported so far for these compounds. The synthesis of these complexes was achieved by addition of three equivalents of nBuLi or NaHMDS (HMDS = N(SiMe3)2) to THF (Li) or Et2O (Na) solutions of 1, thereby yielding the trimetallated derivatives, [M3(S)2L]2 [M = Li, S = THF (3); M = Na, S = Et2O (4)] (Scheme 1), in good yields. X-ray quality crystals of both compounds were obtained from such solutions on cooling (see ESI†).
The molecular structures of complexes 3 and 4 (ESI† and Fig. 1) consist of a distorted face sharing double cubane. Corner-positioned metals are coordinated with three μ3-O atoms and a solvent molecule, completing the archetypal slightly distorted tetrahedral geometry around them [angles O–Mth–O 91.3–132.1° (3), 88.1–143.7° (4)]. In contrast, the metal atoms belonging to the shared face are five-coordinate, with two M–(μ3-O), two M–(μ4-O) and one M–N linkages, defining a square pyramidal geometry with a sum of basal plane angles of 358.9° (3), av. 360.2° (Na), and a range of basal to apical angles of 91.5–95.7° (3) and 79.0–97.8° (4). The corresponding M–O and M–N lengths in complexes 3 and 4 are unexceptional, in fact quite similar to those previously reported for related Li14 or Na15 phenoxide cubanes (M–O), or amine bis(phenolate) complexes (M–N).15e However, both compounds display one significantly elongated M–O bond, 2.227(5) Å (Li3–O3) and 2.530(3) Å (Na8–O4), with these figures being approximately 0.2 and 0.15 Å longer than those typically found in other aryloxide cubanes.14,15 Such an effect might be imposed by the rigid nature of the arms in the tris(phenolate) ligands. Moreover, the corner-positioned metals in these two complexes are further stabilized by the presence of short M⋯C(ipso) and M⋯CH3 intramolecular interactions, which are similar to those previously found by us for simple Li and Na aryloxide complexes supported by polyamine ligands.16 Incidentally, complex 3 seems to be the first lithium phenolate complex displaying a face-sharing double cubane structural motif to be crystallographically characterized. Finally, the available NMR data for complexes 3 and 4 are essentially consistent with the solid-state structures, but support the presence of fluxional processes in solution which exchange the environments of the metals and hydrocarbon chains through open structures (see ESI†).
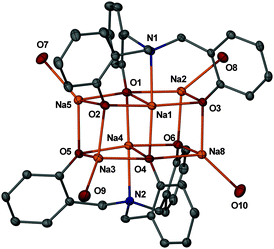 | ||
| Fig. 1 View of the molecular structure of 4 (30% probability) with hydrogens, methyl and ethyl groups omitted. | ||
Keeping in mind that the ligand framework provided by the amine(trisphenol) is flexible enough to accommodate Li and Na cubanes, we turned our attention to the more challenging metallation of the protonated ligand 2;17 this requires the deprotonation of three O–H groups, while leaving the N–H group untouched. Somewhat surprisingly, this was easily accomplished by addition of three equivalents of MHMDS [M = Li and Na] to THF (Li) or Et2O (Na) solutions of 2, with the new tri-metallated complexes (5 and 6 in Scheme 1) then isolated in good yields as crystalline materials and characterized by X-ray crystallography and multinuclear NMR.18
In the solid state, complexes 5 and 6 (Fig. 2 and ESI†) display a zwitterionic ammonium phenolate ligand of approximate C3-symmetry and geometry similar to those previously found in the structures of metal derivatives.9a,19 The M–O distances within these complexes are only marginally shorter than those found for 3 and 4, probably reflecting a less constrained coordination of the tris(phenolate) ligands. The triangle formed by the three metals is capped by the μ3-oxygen atom of the OArCl group, which is less tightly bound. Each metal is further coordinated with one solvent molecule completing a distorted tetrahedral geometry. In addition, we note the presence of short intramolecular M⋯Cl interactions. In fact, the strongest of these interactions in 5 (Li3–Cl5 2.716(2) Å) is only slightly longer than that found for a Li cation coordinated with a haloalkane (2.64 Å).20
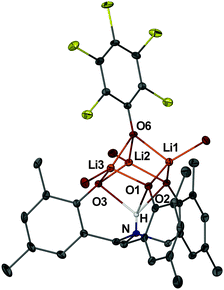 | ||
| Fig. 2 Solid state structure of 5 with most H atoms and solvent molecules (except O atoms) omitted. The central N–H atom was located in the difference Fourier map and refined isotropically. | ||
More intriguing is the role of the central proton which is involved in an unusual trifurcated NH⋯O3 hydrogen bond (mean H(1)–O lengths 2.18 (5) and 2.32 (6) Å). Thus, the cores of these complexes define a distorted cubane motif with the H atoms occupying one of the corner positions (Fig. 3), and therefore effectively replacing one of the metal positions of a typical Li/Na aryloxide cubane. To the best of our knowledge, the structures of complexes 5 and 6 prove for the first time that a hydrogen atom is capable of replacing an alkali metal atom in a typical alkali metal structural motif. In fact, by comparing the cores of complexes 3–6 (Fig. 3) it is clear that the Li/Na to H replacement does not impose significant modifications of the cubane motifs.
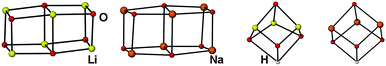 | ||
| Fig. 3 Central cores in complexes 3–6 (from left to right). | ||
Spectroscopic data obtained from solutions of crystalline samples of complexes 5 and 6 are consistent with the retention of “monomeric” cubane structural motifs found in the solid state, and with the pseudo C3-symmetry exhibited by the ligand framework (assuming free rotation for the OArCl group). Thus, the 7Li{1H} NMR spectrum of 5 exhibits only one resonance (δLi = −0.89 ppm), while the 1H NMR spectra of both complexes display just one set of signals for the aromatic protons of the equivalent phenolate rings, along with the expected AB multiplet for three equivalent methylenic (NCH2) groups. The retention of the N–H bond in solution is clearly supported by the appearance of quite deshielded resonance [δH = 12.7 (5), 11.6 ppm (6)] values, which are even higher than that found for the free ligand 2, probably reflecting the existence of stronger NH⋯O3 interactions in these metallated derivatives in solution.
In summary, we have reported for the first time the structural characterization of multi-metallated Group 1 derivatives of amine tris(phenolate) ligands, including the unprecedented participation of a metal-like hydrogen atom in the cubane core of an alkali metal (Li or Na) phenolate. We are now exploring the possibility of extending such a structural analogy to other related systems such as mixed-cubanes with different metals or anions.
We gratefully acknowledge the EPSRC (DTA studentship to D.M.C.), and the European Commission for financial aid (Marie Curie IEF fellowship to D.G.-V., PIEF-2009-235173).
Notes and references
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier, Oxford, 1997 Search PubMed.
- (a) I. F. Silvera and J. W. Cole, J. Phys.: Conf. Ser., 2010, 215, 12194 CrossRef; (b) M. I. Eremets and I. A. Troyan, Nat. Mater., 2011, 10, 927 CrossRef CAS PubMed; (c) F. Hensel and P. P. Edwards, Chem.–Eur. J., 1996, 2, 1201 CrossRef CAS.
- A. N. Rodionov, D. N. Shigorin, T. V. Talalaeva and K. A. Kocheshkov, Dokl. Akad. Nauk SSSR, 1958, 123, 113 CAS.
- P. Kollman, J. Liebman and L. C. Allen, J. Am. Chem. Soc., 1970, 92, 1142 CrossRef CAS.
- (a) S. Scheiner, in Lithium Chemistry: A Theoretical and Experimental Overview, ed. A. M. Sapse and P. Von R. Schleyer, Wiley Interscience, New York, London, Sydney, 1996, ch. 3 Search PubMed; (b) Y. Feng, L. Liu, J.-T. Wang, X.-S. Li and Q.-X. Guo, Chem. Commun., 2004, 88 RSC.
- (a) P. M. Pihko, Angew. Chem., Int. Ed., 2004, 43, 2062 CrossRef CAS PubMed; (b) C. Bolm, T. Rantanen, I. Schiffers and L. Zani, Angew. Chem., Int. Ed., 2005, 44, 1758 CrossRef CAS PubMed; (c) H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924 CrossRef CAS PubMed; (d) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS PubMed.
- (a) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS; (b) C. Pimentel and A. L. McClellan, The Hydrogen Bond, Freeman, San Francisco, 1960 Search PubMed; (c) F. Renaud, C. Piguet, G. Bernardinelli, J.-C. G. Bünzli and G. Hopfgartner, J. Am. Chem. Soc., 1999, 121, 9326 CrossRef CAS.
- For some reviews in the structural chemistry of organocomplexes of alkali metals see: (a) W. N. Setzer and P. von R. Schleyer, Adv. Organomet. Chem., 1985, 24, 353 CrossRef CAS; (b) C. Schade and P. von R. Schleyer, Adv. Organomet. Chem., 1987, 27, 169 CrossRef CAS; (c) R. E. Mulvey, Chem. Soc. Rev., 1991, 20, 167 RSC; (d) Z. Rappoport and I. Marek, The Chemistry of Organolithium Compounds, Wiley, New York, 2004 Search PubMed; (e) R. A. Gossage, J. T. B. H. Jastrzebski and G. Van Koten, Angew. Chem., Int. Ed., 2005, 44, 1448 CrossRef CAS PubMed.
- (a) M. G. Davidson, C. L. Doherty, A. L. Johnson and M. F. Mahon, Chem. Commun., 2003, 1832 RSC; (b) S. D. Bull, M. G. Davidson, A. L. Johnson, M. R. Mahon and D. E. J. E. Robinson, Chem.–Asian J., 2010, 5, 612 CrossRef CAS PubMed; (c) A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 1293 RSC; (d) A. J. Chmura, C. J. Chuck, M. G. Davidson, M. D. Jones, M. D. Lunn, S. D. Bull and M. F. Mahon, Angew. Chem., Int. Ed., 2007, 46, 2280 CrossRef CAS PubMed.
- (a) K. Ruhlandt-Senge, K. W. Henderson and P. C. Andrews, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and M. Mingos, 2007, ch. 1, vol. 2 Search PubMed; (b) F. Pauer and P. P. Power, in Lithium Chemistry, ed. A. M. Sapse and P. Von R. Schleyer, Wiley Interscience, New York, London, Sydney, 1996, p. 295 Search PubMed; (c) D. C. Bradley, R. C. Mehrotra, I. P. Rothwell and A. Singh, in Alkoxo and Aryloxo Derivatives of Metals, Academic Press, London, 2001 Search PubMed.
- M. Kol, M. Shamis, I. Goldberg, Z. Goldshmit, S. Alfi and E. Hayut-Salant, Inorg. Chem. Commun., 2001, 4, 177 CrossRef CAS.
- W. Huang, Z. Chu and F. Xu, J. Mol. Struct., 2008, 885, 154 CrossRef CAS.
- (a) L. Michalczyk, S. de Gala and J. W. Bruno, Organometallics, 2001, 20, 5547 CrossRef CAS; (b) C. J. Whiteoak, B. Gjoka, E. Martin, M. M. Belmonte, E. C. Escudero-Adán, C. Zonta, G. Licini and A. W. Kleij, Inorg. Chem., 2012, 51, 10639 CrossRef CAS PubMed.
- T. J. Boyle, D. M. Pedrotty, T. M. Alam, S. C. Vick and M. A. Rodriguez, Inorg. Chem., 2000, 39, 5133 CrossRef CAS PubMed.
- See for example: (a) P. A. Van der Schaaf, J. T. B. H. Jastrzebski, M. P. Hogerheide, W. J. J. Smeets, A. L. Spek, J. Boersma and G. Van Koten, Inorg. Chem., 1993, 32, 4111 CrossRef CAS; (b) W. J. Evans, R. E. Golden and J. W. Ziller, Inorg. Chem., 1993, 32, 3041 CrossRef CAS; (c) M. Kunert, E. Dinjus, M. Nauck and J. Sieler, Chem. Ber., 1997, 130, 1461 CrossRef CAS; (d) D. J. McDougall, B. C. Noll and K. W. Henderson, Inorg. Chem., 2005, 44, 1181 CrossRef PubMed; (e) W. Clegg, M. G. Davidson, D. V. Graham, G. Griffen, M. D. Jones, A. R. Kennedy, C. T. O'Hara, L. Russo and C. M. Thomson, Dalton Trans., 2008, 1295 RSC.
- (a) D. M. Cousins, M. G. Davidson, D. García-Vivó and M. F. Mahon, Dalton Trans., 2010, 39, 8203 RSC; (b) B. Calvo, M. G. Davidson and D. García-Vivó, Inorg. Chem., 2011, 50, 3589 CrossRef CAS PubMed.
- A preferential N–H deprotonation would be anticipated in view of the pKa values reported for related species [pKa = 9.80 (Me3NH+), 10.86 (2,4,6-trimethylphenol)].
- For 6 all the crystallizations yielded low-quality crystals which led to poor diffraction data. However, the crystallographic study allows for an unambiguous characterization of the structural motif.
- (a) A. J. Nielson, C. Shen and J. M. Waters, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, m494 CrossRef; (b) J. D. Chartres, A. Dahir, P. A. Tasker and F. J. White, Inorg. Chem. Commun., 2007, 10, 1154 CrossRef CAS.
- L. O. Müller, R. Scopelitti and I. Krossing, Z. Anorg. Allg. Chem., 2008, 634, 1035 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, including spectroscopic data for new compounds. CCDC 916821–916824. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc47393g |
| This journal is © The Royal Society of Chemistry 2013 |