 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A self-switchable Ag nanoreactor exhibiting outstanding catalytic properties†
Shunsheng
Cao
*ab,
Juanrong
Chen
b,
Yi
Ge
b,
Long
Fang
a,
Ying
Zhang
a and
Anthony P. F.
Turner
*c
aSchool of Materials Science and Engineering, Jiangsu University, Zhenjiang, 212013, P. R. China. E-mail: sscaochem@hotmail.com
bCranfield Health, Cranfield University, Bedfordshire, MK43 0AL, UK
cBiosensors & Bioelectronics Centre, IFM, Linköping University, SE-58183, Linköping, Sweden. E-mail: Anthony.turner@liu.se
First published on 24th October 2013
Abstract
A highly efficient nanoreactor that contains silver nanoparticles in hollow silica spheres and an interpolymer network as a gate-keeper has been developed following a facile procedure. The fast “signal-triggered” switch of the smart network results in a high reactivity and a high response rate, yielding improved potential for many practical applications.
An emerging trend in nanotechnology and advanced materials is the design of controllable or “signal-triggered” metal nanoparticles with the objective of creating “stimuli-responsive” nanoreactors.1 Free metal nanoparticles tend to aggregate and are hence difficult to incorporate into tunable or switchable configurations. To overcome this limitation, carrier systems have been constructed to immobilize and stabilise such nanoparticles.2 An ideal design by combining metal nanoparticles with advanced materials can offer materials with new functions, such as modulated catalysis.3 By employing the unique thermo-sensitive phase transition of PNIPA,4 catalysts or substrates bound within PNIPA can be readily manipulated by virtue of the reversible swelling and shrinking of PNIPA chains, which are induced by the tunable hydrogen bonding interactions surrounding them.5 Recently, Lu et al.6 have reported an Au–PNIPA yolk–shell system. They found that such a PNIPA-based nanoreactor showed a high conversion of 4-nitrophenol, while the transition status of hydrophilicity is presented at relatively low temperature. After their phase becomes hydrophobic with increased external temperature, the polymeric nanomatrices become insoluble in water, thereby causing a sharp decrease in the amount of reduced 4-NP. Clearly, in most cases such nanoreactors with inverse response do not have high potential for practical applications, simply due to the low reactivity and responsive kinetics at low temperature.7 By contrast, the construction of a nanocatalyst with a positive responsive function, in which a given reaction is induced by an increase rather than a decrease in temperature, would generate widespread applications such as catalysis and sensing. Therefore, novel designs and strategies for creating nanoreactors with positive response are urged.
In the case of supported metal nanostructures, a highly efficient method is to involve small clusters or particles of metal with high surface areas loaded in inert hollow spheres, or the so-called yolk–shell or rattle-type particles, because they can be used as storage spaces or reaction chambers while supplying the necessary paths for the design of modulated catalysis.8 Among them, hollow silica spheres have become the subject of intense research due to their distinct characteristics including high surface area, tunable porous shell, high mechanical/thermal stability and facile modification.9 More importantly, their micro/meso-pores in the walls allow for the transport of reactant and product molecules through the shell,10 thus providing a confined microenvironment for chemical reactions.9,11 The local concentration of reactants in the hollow sphere can be higher than that in the surrounding environment due to the adsorption of reactants in the porous wall, which leads to an improved reaction rate.11 Therefore, this material appears to be especially attractive to modulate the catalytic activity of the loaded metal nanoparticles via a functionalized architecture of the wall.
Building on these previous studies, we explored the idea of creating a highly efficient and positively thermo-sensitive Ag nanoreactor with tunable catalytic activity, in which hollow silica spheres were used as hosts for the immobilization of Ag nanoparticles and temperature was used as a trigger for a catalytic reaction. To fulfill this requirement we have chosen PNIPA and PAMPS as the thermosensitive components because they can offer a faster “signal-triggered” switch than pure PNIPA-based materials. The proposed Ag nanoreactor exhibited positively responsive characteristics and faster response kinetics, because the catalytic behavior of the nanoreactor was induced by an increase rather than a decrease in temperature. This self-switchable nanoreactor not only offered an advantage over the negatively thermo-responsive switches used in pure PNIPA-based nanoreactors, but also achieved tunable catalytic activity and a higher degree of change in the catalytic activity after the external trigger was applied, which allowed better control of reactants accessing the loaded Ag nanoparticles.7 In this communication, we demonstrated that such a responsive nanoreactor could be engineered by combining hollow silica spheres, Ag nanoparticles, and a PNIPA/PAMPS smart interpenetrating network. Scheme 1 illustrates the underlying principle of this system: Ag nanoparticles were loaded in hollow silica spheres with the thermo-sensitive PNIPA/PAMPS network. The entrance of this shell network could be switched “on” or “off” in a well-defined manner by changing the external temperature. We verified this potential and the efficacy as an Ag nanoreactor by measuring the reduction of 4-NP via sodium borohydride.12 The self-switchable nanoreactor exploited a positively thermo-sensitive effect, higher relative reactivity, and modulated catalysis, yielding improved potential for many practical applications.7
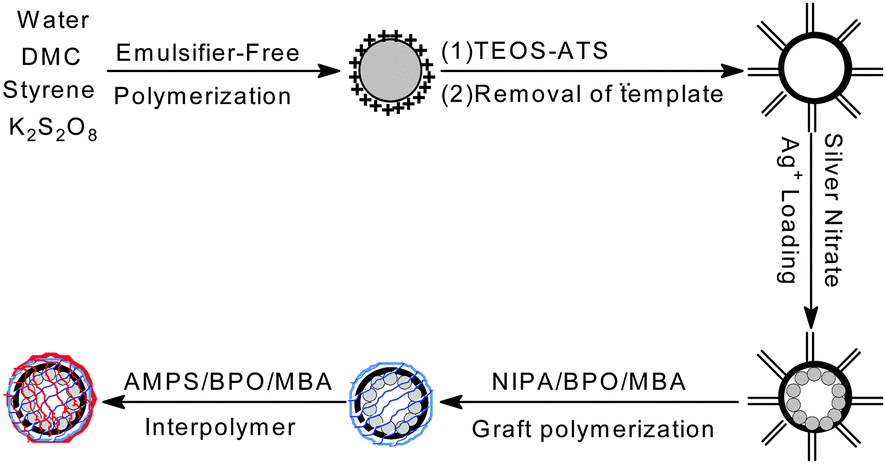 | ||
| Scheme 1 Construction of the self-switchable Ag nanoreactors. | ||
Ag–N–A was synthesized in five steps. Firstly, CPS was prepared according to our previous studies.13 Secondly, the functional HS was developed by using CPS as the template. Thirdly, Ag+ was loaded in the HS via electrostatic interaction. Unloaded Ag+ was removed by dialysis against deionized water, preventing free silver nanoparticles from extensive aggregation.14 TEM and SEM images indicated that, after being washed several times, the prepared Ag–HS had a well-defined morphology. Distribution studies of Ag nanoparticles showed that HS effectively prevented the formation of free Ag nanoparticles in the bulk phase, resulting in a very small size (average value: 8.52 nm). The content of Ag nanoparticles in the hollow silica measured by ICP-AES was about 1.362% (w/w). In addition, the elemental mapping analysis (STEM-EDX) further confirmed that Ag was almost homogeneously distributed over the entire particle, generating a good dispersion. After the Ag was loaded, NIPA was added and grafted onto the surface of the hollow silica spheres, and then AMPS, in a stoichiometric ratio with NIPA, was used to swell the beads and was then polymerized in the presence of MBA, a cross-linker, onto the surface of the HS–PNIPA to form an interpolymer network. Finally, the resulting PNIPA/PAMPS was reduced to Ag nanoparticles with an excess amount of sodium borohydride to produce a self-switchable Ag nanoreactor. The formation of the interpolymer shell was confirmed by TEM. Graft polymerization of PNIPA/PAMPS gave a uniform thin layer surrounding the outer surface of the hollow silica spheres, resulting in an improved wall thickness in comparison to functional HS. DLS measurement further confirmed that the morphology and dispersity of the Ag–N–A exhibited no obvious damage before and after the graft polymerization of PNIPA/PAMPS. In addition, if the porous shell of HS could be effectively blocked by the interpolymer, it should follow that the Ag–N–A presented lower N2 adsorption and BET surface area in comparison to HS. BET measurements indicated that the amount of N2 adsorbed and the surface area of Ag–N–A dropped sharply from 145.21 cc g−1 to 32.49 cc g−1 and from 72.589 m2 g−1 to 19.267 m2 g−1, respectively, suggesting that the PNIPA/PAMPS effectively covered the porous shell of hollow silica spheres.
UV/Vis spectra were obtained to measure the amount of reduced 4-NP by observing the peak at 400 nm, which is attributed to the 4-nitrophenate ions produced during the catalytic reduction of 4-NP.3,12 As shown in Fig. 1, below the LCST (32 °C),15 a low conversion of 4-NP and slow increase were observed, indicating that the entrance of the Ag–N–A nanoreactor was largely blocked due to the coalescence of PNIPA and PAMPS branches via hydrogen bonding interactions. Moreover, the prepared Ag–N–A exhibited good reusability and stability because its morphology and structure exhibit no obvious damage after catalytic reduction of 4-NP (three times), which was vital for many practical applications. Interestingly, a dramatically increased amount of reduced 4-NP was obtained at 38 °C, indicating that 4-NP could freely access the Ag–N–A because the hydrogen bonding was subverted above the LCST.16 This temperature range is in agreement with the thermo-sensitive region of the PNIPA-based polymer discussed earlier, suggesting that the association and dissociation between PNIPA and PAMPS played a decisive role in creating self-switchable and positively thermo-sensitive effects for the Ag–N–A nanoreactor.
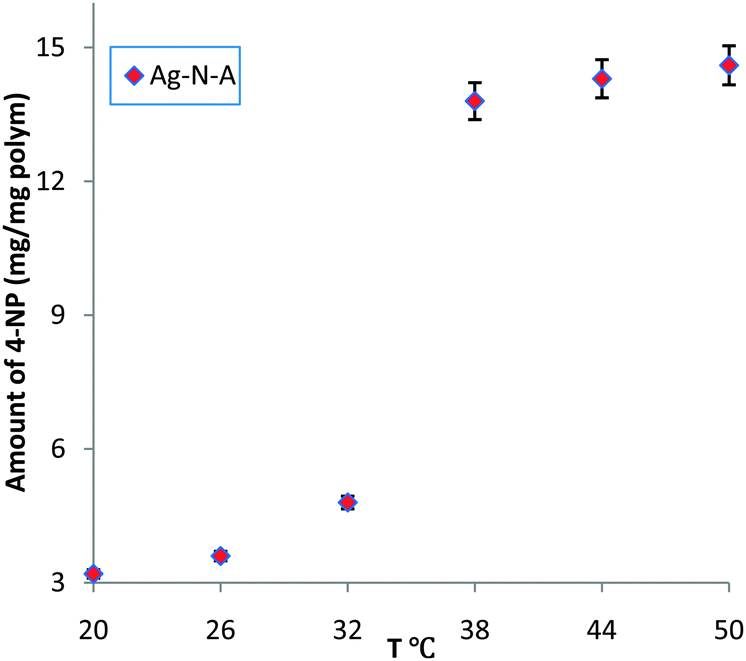 | ||
| Fig. 1 Catalytic activity of the prepared Ag–N–A for the reduction of 4-NP. | ||
In order to further study modulated catalysis and to detail relative reactivity, we prepared two typical nanoreactors (Ag–HS and Ag–N) and compared their relative reactivities with Ag–N–A. Over the whole temperature range (20–50 °C), Fig. 2a, there was no sharp increase in the amount of reduced 4-NP for Ag–HS, suggesting a stepwise mode. Unlike Ag–HS, both Ag–N and Ag–N–A exhibited marked increases in the amount of reduced 4-NP at 38 °C, as shown in Fig. 2b, indicating a tunable model. Furthermore, the Ag–N and Ag–N–A offered higher relative reactivity than Ag–HS (1.37 times). In particular, the synthesized Ag–N–A (2.725 times) delivered a higher degree of change in catalytic activity and a higher response rate than Ag–N (2.243 times), indicating that Ag–N–A could effectively overcome the disadvantage of the low response rate seen with pure PNIPA, which usually requires a long time to reach equilibrium.7 A higher activity of Ag–N–A was observed below the LCST, in contrast to the Ag–N nanoreactor, which might be related to the difference in the relative hydrophilicity/hydrophobicity of AMPS and NIPA.17
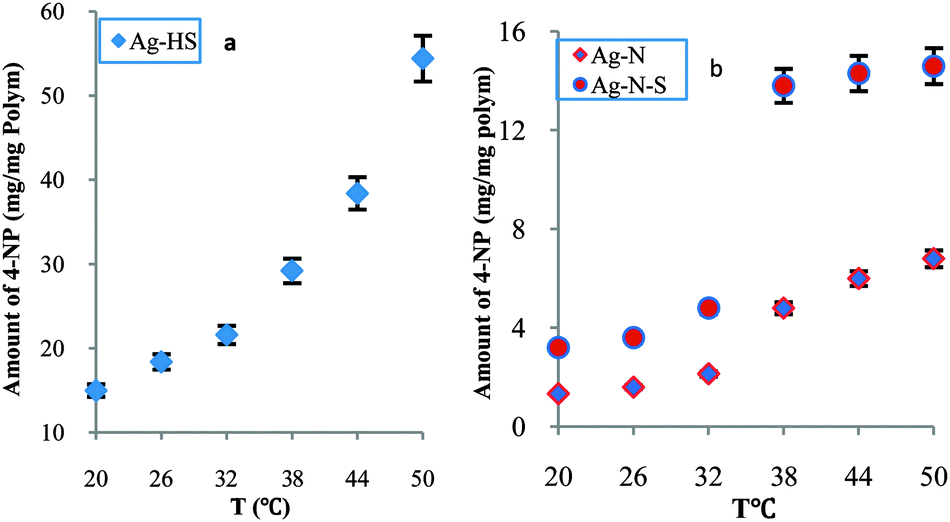 | ||
| Fig. 2 Relative reactivity of Ag–HS (a) and of Ag–N–A and Ag–N (b). | ||
The key objective is the construction of nanocatalysts with strong and tunable chemical activity, stability, and easy recovery.18 In this work, the density of Ag nanoparticles within HS could be further tuned by simply varying the initial loading content of AgNO3 due to the high surface area of HS. Clearly, a higher amount of reduced 4-NP was obtained with higher initial loading of AgNO3. Interestingly, the degree of permeation between 32 and 38 °C could be further increased with the increase in the initial loading of AgNO3 as shown in Fig. 3. With 0.2 g of AgNO3, the degree of change increased to about 1.9 times of the original rate. When the amount of AgNO3 approached 0.4 g, the degree of change was about 2.3 times. The highest degree of change (about 2.9 times) was achieved with 1.0 g of AgNO3. Understandably, it was not easy to further increase such a degree of change by simply adding more AgNO3 due to the occurrence of Ag leaching, indicating that Ag nanoparticles generally loaded in HS were exposed to PNIPA/PAMPS chains. This change in Ag loading permitted more Ag nanoparticles to access 4-NP even below the LCST, resulting in a higher amount of reduced 4-NP, whilst decreasing the degree of change.
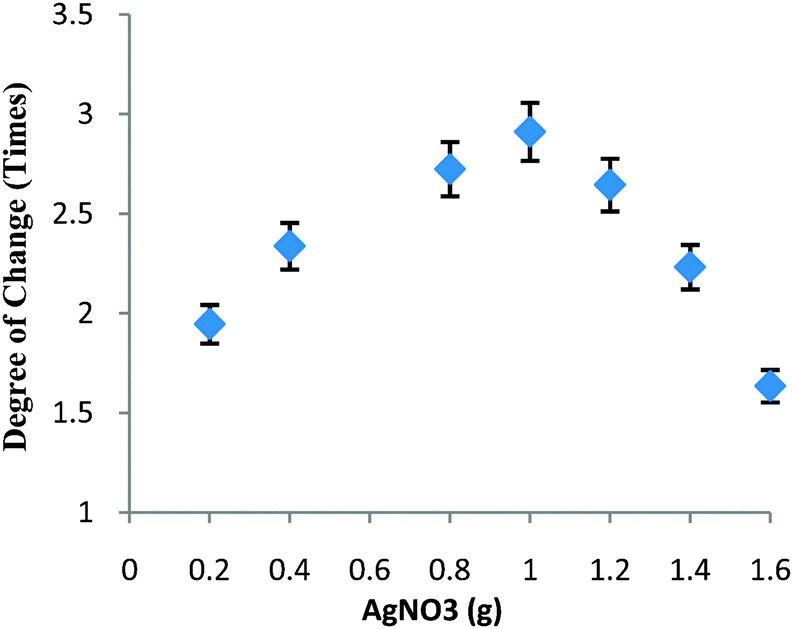 | ||
| Fig. 3 Degree of change in Ag–N–A nanoreactors between 32 and 38 °C. | ||
A defective network could cause steric mismatch between PNIPA and PAMPS, resulting in discordant association and dissociation. Only the interpolymer network could construct the complete smart association and dissociation, and thereby the best thermo-sensitive responsiveness was achieved. To monitor this interaction, the effect of MBA on the catalytic behaviour of Ag–N–A was investigated. The results showed that MBA played a very important role in creating modulated catalysis for Ag–N–A. When the amount of MBA added was 0.5%, the Ag–N–A offered tunable catalysis as aforedescribed. When the amount of MBA was increased to 1%, the similar catalytic behavior was observed, although the amount of reduced 4-NP was greatly decreased due to the presence of a highly cross-linked network. In the absence of MBA, the Ag–N–A showed disordered catalytic properties due to random association and dissociation.
In summary, we introduced a self-switchable Ag nanoreactor, in which hollow silica spheres were used as hosts for the immobilization of Ag nanoparticles and a thermo-sensitive interpolymer PNIPA/PAMPS could act as a “gate-keeper” that can be maneuvered by a temperature change. These hybrid materials combined the advantages of hollow silica spheres, Ag nanoreactors, and tunable catalytic activities, resulting in a fast response rate and highly efficient catalysts for the reduction of 4-NP in aqueous solution. Although hollow silica spheres and an interpolymer have been chosen as a basis of this demonstration, the design principles and synthetic methods are applicable to a wide variety of porous structures and polymers or molecules that are sensitive to external stimuli. Therefore, the advanced properties of the self-switchable Ag–N–A system have considerable potential to open a new avenue for tailoring the catalytic activity of metal nanoparticles toward a given reaction.
The authors thank the Research Directorate-General of the European Commission for financial support to carry out this work under the framework of Marie Curie Action FP7-People-IIF-2010 (No. 275336) awarded to Shunsheng Cao.
Notes and references
- (a) F. Y. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC; (b) M. Zahmakiran and S. Ozkar, Nanoscale, 2011, 3, 3462–3481 RSC.
- L. A. Lyon, Z. Meng, N. Singh, C. D. Sorrell and A. St. John, Chem. Soc. Rev., 2009, 38, 865–874 RSC.
- (a) S. W. Choi, Y. Zhang and Y. Xia, Angew. Chem., Int. Ed., 2010, 49, 7904–7908 CrossRef CAS PubMed; (b) Y. Lu and M. Ballauff, Prog. Polym. Sci, 2011, 36, 767–792 CrossRef CAS PubMed.
- (a) Y. M. Yamada, S. M. Sarkar and Y. Uozumi, J. Am. Chem. Soc., 2012, 134, 3190–3198 CrossRef CAS PubMed; (b) S. Li, Y. Ge and A. P. F. Turner, Adv. Funct. Mater., 2011, 21, 1194–1200 CrossRef CAS.
- (a) G. Qing and T. Sun, Adv. Mater, 2011, 23, 1615–1620 CrossRef CAS PubMed; (b) S. Li, Y. Ge, A. Tiwari and S. Cao, Small, 2010, 6, 2453–2459 CrossRef CAS PubMed.
- S. Wu, J. Dzubiella, J. Kaiser, M. Drechsler, X. Guo, M. Ballauff and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 2229–2233 CrossRef CAS PubMed.
- (a) W. Xu, X. Yin, G. He, J. Zhao and H. Wang, Soft Matter, 2012, 8, 3105–3111 RSC; (b) J. A. Crowe and J. Genzer, J. Am. Chem. Soc., 2005, 127, 17610–17611 CrossRef CAS PubMed; (c) Y. Dou, J. Han, T. Wang, M. Wei, D. G. Evans and X. Duan, Langmuir, 2012, 28, 9535–9542 CrossRef CAS PubMed.
- (a) J. Lian, Y. Xu, M. Lin and Y. Chan, J. Am. Chem. Soc., 2012, 134, 8754–8757 CrossRef CAS PubMed; (b) M. Sanlés-Sobrido, M. Pérez-Lorenzo, B. Rodríguez-González, V. Salgueiriño and M. A. Correa-Duarte, Angew. Chem., Int. Ed., 2012, 51, 3877–3882 CrossRef PubMed; (c) C. Aydin, J. Lu, N. D. Browning and B. C. Gates, Angew. Chem., Int. Ed., 2012, 51, 5929–5934 CrossRef CAS PubMed.
- (a) M. Xiao, C. Zhao, H. Chen, B. Yang and J. Wang, Adv. Funct. Mater., 2012, 22, 4526–4532 CrossRef CAS; (b) J. Hu, M. Chen, X. Fang and L. Wu, Chem. Soc. Rev., 2011, 40, 5472–5491 RSC.
- (a) P. Chung, R. Kumar, M. Pruski and V. S. Y. Lin, Adv. Funct. Mater., 2008, 18, 1390–1398 CrossRef CAS; (b) X. Du, L. Yao and J. He, Chem.–Eur. J., 2012, 18, 7878–7885 CrossRef CAS PubMed.
- (a) J. Han, Y. Liu and R. Guo, Adv. Funct. Mater., 2009, 19, 1112–1117 CrossRef CAS; (b) Y. Yamada, M. Mizutani, T. Nakamura and K. Yano, Chem. Mater., 2010, 22, 1695–1703 CrossRef CAS; (c) S. M. Leeder and M. R. Gagné, J. Am. Chem. Soc., 2003, 125, 9048–9054 CrossRef CAS PubMed.
- (a) C. Yuan, W. Luo, L. Zhong, H. Deng, J. Liu, Y. Xu and L. Dai, Angew. Chem., Int. Ed., 2011, 50, 3515–3519 CrossRef CAS PubMed; (b) X. Zhang and Z. Su, Adv. Mater., 2012, 24, 4574–4577 CrossRef CAS PubMed.
- (a) S. Cao, L. Fang, Z. Zhao, Y. Ge, S. Piletsky and A. P. F. Turner, Adv. Funct. Mater., 2013, 23, 2162–2167 CrossRef CAS; (b) S. Cao, Z. Zhao, X. Jin, W. Sheng, S. Li, Y. Ge, M. Dong and L. Fang, J. Mater. Chem, 2011, 21, 19124–19131 RSC.
- T. Wu, Z. Ge and S. Liu, Chem. Mater., 2011, 23, 2370–2380 CrossRef CAS.
- N. Yan, J. Zhang, Y. Yuan, G. T. Chen, P. J. Dyson, Z. C. Li and Y. Kou, Chem. Commun., 2010, 46, 1631–1633 RSC.
- (a) Z. Ge, D. Xie, D. Chen, X. Jiang, Y. Zhang, H. Liu and S. Liu, Macromolecules, 2007, 40, 3538–3546 CrossRef CAS; (b) S. Song and N. Hu, J. Phys. Chem. B, 2010, 114, 5940–5945 CrossRef CAS PubMed.
- H. Tokuyama, M. Sasaki and S. Sakohara, Colloids Surf., A, 2006, 273, 70–74 CrossRef CAS PubMed.
- Y. Zhang, X. Han, R. Liu, Y. Liu, H. Huang, J. Zhang, H. Yu and Z. Kang, J. Phys. Chem. C, 2012, 116, 20363–20367 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc47361a |
| This journal is © The Royal Society of Chemistry 2014 |