 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral resolution through stereoselective transglycosylation by sucrose phosphorylase: application to the synthesis of a new biomimetic compatible solute, (R)-2-O-α-D-glucopyranosyl glyceric acid amide†
Patricia
Wildberger
a,
Lothar
Brecker
b and
Bernd
Nidetzky
*a
aInstitute of Biotechnology and Biochemical Engineering, Graz University of Technology, Petersgasse 12/1, A-8010 Graz, Austria. E-mail: bernd.nidetzky@tugraz.at
bInstitute of Organic Chemistry, University of Vienna, Währingerstraße 38, A-1090 Vienna, Austria
First published on 30th October 2013
Abstract
Sucrose phosphorylase catalysed glycosylation of glyceric acid amide with complete regio- and diastereo-selectivity is studied. (R)-2-O-α-D-Glucopyranosyl glyceric acid amide was obtained in high yield from single-step transformation of racemic glyceric acid amide and sucrose. Non-productive binding of (S)-glyceric acid amide appeared to underlie strict enantiodiscrimination by the enzyme, thus supporting chiral resolutions based on stereoselective transglycosylation.
Biocatalytic transglycosylations constitute highly expedient transformations in synthetic organic chemistry. Transglycosylations can be applied broadly to the targeted modification of small molecules, polymers and even materials through covalent attachment of sugar moieties.1–4 Chemically, transglycosylations proceed via transfer of a glycosyl residue from a donor glycoside onto a suitable functional group, typically an alcohol, on an acceptor substrate (Scheme 1). Biocatalytic transglycosylations usually involve robust and co-substrate-independent enzymes that utilise inexpensive donor substrates. They are therefore regarded as practical methodology of glycoside synthesis that can be applied readily across scales. A number of glycosidic products prepared through biocatalytic transglycosylations have important industrial applications in the food, cosmetic and fine-chemical sectors.5,6 Despite these clear advantages, however, there exists a significant gap in the scope of biocatalytic transglycosylations, in that known enzymes often lack acceptor substrate regioselectivity, stereoselectivity, or both.7 Site-heterogeneous glycosylation of acceptor substrates offering more than a single reactive group severely restricts practicability of the method. Also, chiral resolution of racemic alcohols through diastereoselective glycosylation has not so far been a useful option for biocatalytic synthesis.
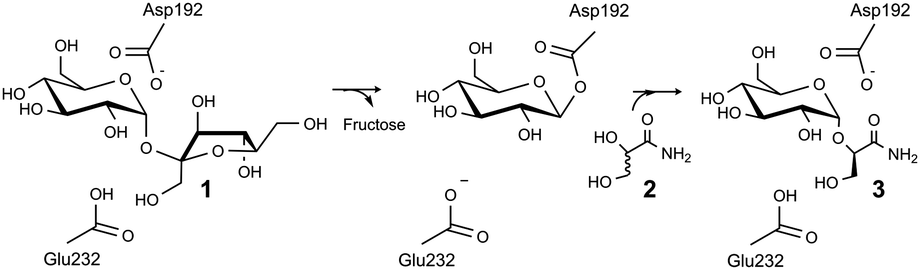 | ||
| Scheme 1 Proposed mechanism of sucrose phosphorylase for kinetic resolution of 2 and chiral synthesis of 3. | ||
We herein provide a first-time example of enzymatic transglycosylation with complete regio- and stereo-selectivity. When using sucrose (1) as a glucosyl donor and racemic glyceric acid amide (2) as an acceptor (Scheme 1), sucrose phosphorylase was highly specific for formation of (R)-2-O-α-D-glucopyranosyl glyceric acid amide (3) as a single diastereomerically pure glucosyl transfer product. The enzymatic reaction thus supports different chiral resolution strategies for 2 in which synthesis of a structurally and stereochemically well-defined glycoside 3 or recovery of an unreacted acceptor (S enantiomer of 2) is targeted. Based on the results of molecular docking analysis we explain how sucrose phosphorylase achieves strict discrimination between R and S enantiomers of 2. We also provide evidence of the role of the acceptor's 1,2-diol group for substrate binding recognition and positioning for regio- and stereo-selective catalytic glycosylation. Our findings suggest that sucrose phosphorylase-catalysed chiral synthesis from racemic diols (for which 2 is a representative example) could be an interesting novel strategy for application of enzymatic transglycosylations in organic synthesis. The work of Franssen and co-workers has recently shown that the enzyme promotes 2-O-α-D-glucosylation of various diols with useful stereoselectivity.8 Glycoside 3 was synthesised herein for the first time. It is a close structural analogue of (R)-2-O-α-D-mannopyranosyl glyceric acid amide (4), also called Firoin-A, which was isolated from the thermophilic bacterium Rhodothermus marinus. Firoin-A is an intracellular compatible solute that the organism accumulates in response to a high salt environment. Firoin-A is described as a useful stabiliser of proteins in different applications, refolding processes in particular.9
Sucrose phosphorylase promotes transglycosylation from sucrose in a two-step catalytic mechanism, depicted in Scheme 1, that involves formation of a β-glucosyl enzyme intermediate.10 In the natural reaction of the enzyme, phosphate is the terminal glucosyl acceptor and α-D-glucose-1-phosphate is formed. In the absence of phosphate, when an alternative acceptor alcohol is present, a new O-α-D-glycoside is synthesised, provided that the acceptor competes effectively with water for reaction with the enzyme intermediate. Acceptor 2 was synthesised chemically as described in the ESI† (Materials). Purified sucrose phosphorylase from Leuconostoc mesenteroides, recombinantly produced in Escherichia coli as described in the ESI† (Enzyme production), was used. We incubated a mixture of 1 and 2 (each 500 mM) in the presence of an enzyme (30 units of activity per mL; see the ESI† for methods and reaction conditions used) and analysed samples from the reaction mixture using liquid chromatography. Release of fructose from conversion of 1 exceeded by up to 2-fold the concurrent formation of glucose, providing indirect evidence for the occurrence of transglycosylation next to a significant hydrolysis of the enzyme intermediate. The product mixture in which about 45–50% of 2 has been consumed was analysed by NMR, and Fig. 1 summarises pertinent results. The glucosylation of 2 was confirmed and it was shown that only a single main transglycosylation product has been formed in the enzymatic reaction. Identity of the fully stable product was established as 2-O-α-D-glucopyranosyl glyceric acid amide. The interglycosidic linkage was identified by the 3JH–C couplings determined from HMBC spectra showing interactions between the H-1 of the glycoside and C-2′ of the aglycon, as well as between the C-1 of the glycoside and H-2′ of the aglycon. Furthermore, the two protons in these positions have an intensive dipolar interaction, detected in the 2D NOESY spectrum. The α-anomeric form of the glucopyranosyl moiety was determined from the typical 13C and 1H chemical shifts as well as from the small coupling constant (3.8 Hz) between the protons in positions one and two of the glycoside (Fig. 1). Although the absolute configuration of C-2′ could not be elucidated from the NMR data, the absence of signal splitting of H-1 of the glycoside and H-2′ of the aglycon strongly hinted the presence of a diastereomerically pure glycoside (d.e. ≥99%). A racemic mixture would have led to a double set of signals with slightly different chemical shifts (ESI,† Fig. S1).
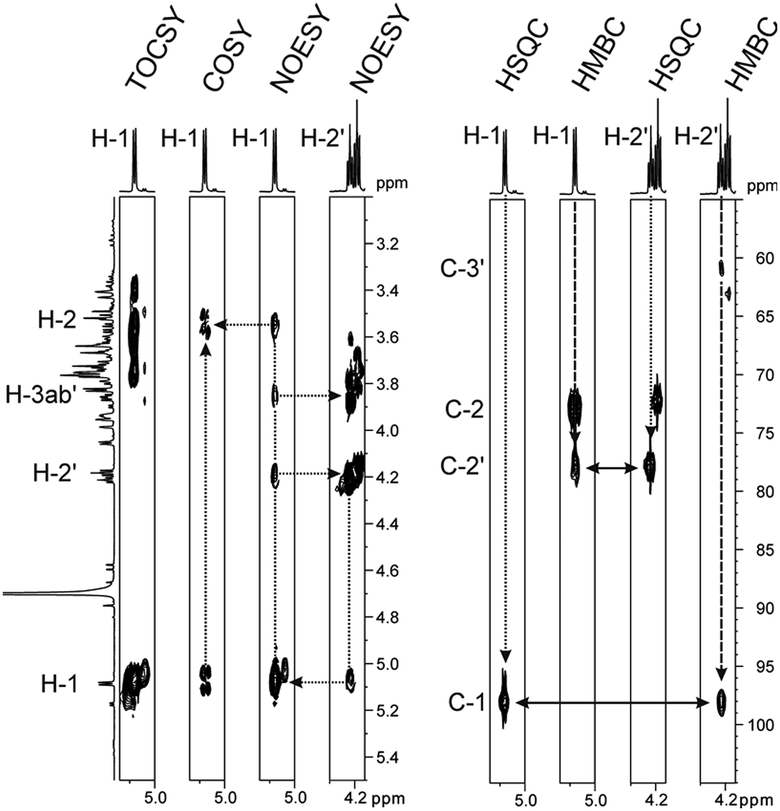 | ||
| Fig. 1 Details of 2D NMR spectra recorded from the reaction mixture to determine the product's glycosidic linkage. TOCSY and COSY traces of the anomeric proton H-1 allow for determination of further protons in the glucosidic moiety. The NOESY trace indicates spatial closeness of H-1 to H-2′ and H-3ab′ of the glyceric acid amide aglycon. The concomitant trace of H-2′ is shown. HSQC and HMBC traces indicate the 1JH–C and 3JH–C couplings of H-1 and H-2′, respectively. The long-range coupling over three bonds clearly indicates the glycosidic bond to be formed with the secondary alcohol of the glyceric acid amide. The signal group directly neighboured to those of H-2′ belongs to the H-2 of the not transformed substrate. | ||
In the next step, we applied chiroptical techniques for the determination of the transglycosylation product's absolute configuration. Due to unavailable reference data for the product, an indirect approach was chosen. Residual 2 was recovered from the reaction mixture as described in the ESI† (conversion of 2 ≥35%) and the optical rotation [α]22D of the sample was determined to be +15°. The significant sign of the measurement can be compared with the literature where an optical rotation of [α]20D = −63° was reported for (R)-2.11 A circular dichroism spectrum of the sample (ESI,† Fig. S2) showed only one extremum at λ = 230 nm with Δε = +15.7. These data can be compared to circular dichroism spectra of (R) and (S)-serine because serine shares some structural similarity with 2. The S enantiomer of serine shows a similar positive circular dichroism band as the purified residual sample whereas the circular dichroism band of (R)-serine is negative.12,13 In summary, therefore, strong evidence is presented supporting the suggestion that 2 recovered from the reaction with sucrose phosphorylase has S configuration. Stereochemical structure of the glucosylation product is assigned accordingly as (R)-2-O-α-D-glucopyranosyl glyceric acid amide (3). Synthesis of pure 3 necessitates that sucrose phosphorylase provides complete regio- and diastereo-selectivity in the reaction with 2 which is a combination of selectivity properties not previously observed in other transglycosidase-catalysed conversions. Remaining 2 was recovered in excellent yield from the reaction (>40%), suggesting the possibility of kinetic resolution via stereoselective transglycosylation.
To gain a molecular interpretation of the phosphorylase's remarkable selectivity, we performed energy-minimised docking studies on the high-resolution crystal structure of the enzyme (from Bifidobacterium adolescentis)10 whereby the R and S enantiomers of 2 were placed flexibly in the acceptor-binding pocket of the β-glucosyl enzyme intermediate. Note: active-site residues of sucrose phosphorylase from B. adolescentis are superimposable on the homology model of the enzyme from L. mesenteroides (ESI,† Fig. S5). Fig. 2 illustrates the obtained best-fit binding modes for (R)-2 and (S)-2. Reactive group arrangement was highly plausible for the (R)-2 pose where the 2-OH was well aligned for nucleophilic attack on the glucosyl anomeric carbon, catalytically assisted by Glu-232 functioning as a general base, consistent with the proposed catalytic mechanism of the enzyme. The primary hydroxyl of (R)-2 was coordinated tightly by Asp-290 and Gln-345. The amide group of (R)-2 was bound weakly by comparison. For (S)-2 the predicted binding mode was characterised by a conspicuously large distance between the reactive carbon and oxygen, clearly indicating that the (S)-2 pose was non-productive for reaction. As in (R)-2, however, the primary hydroxyl of (S)-2 was accommodated through hydrogen bonding interactions with Asp-290 and Gln-345. Other acceptors harbouring a terminal diol moiety (Fig. 3) were previously shown to be glucosylated by the enzyme with useful stereoselectivity, resulting in 2-O-α-D-glucosidic products of high diastereomeric purity.8,14 Molecular docking of (R)- and (S)-forms of glyceric acid and 3-methoxy-1,2-propanediol to sucrose phosphorylase underpins the observed preference for reaction with one enantiomer of the acceptor, suggesting this to be the (R)-form in each case (ESI,† Fig. S3). In search of an interpretable structural parameter of enzyme stereoselectivity in reactions with different acceptors, we compared reactive atom distances (glucosyl carbon and acceptor oxygen; C1⋯O2) in the respective (R)- and (S)-acceptor poses. The C1⋯O2 distance in the (S)-acceptor pose was highly variable across the different acceptors used (ESI,† Fig. S3), while the corresponding C1⋯O2 distance in the (R)-acceptor pose was constant at about 3.0 Å. The ratio of the C1⋯O2 distance in the (S)- and (R)-acceptor pose appears to correlate with the observed diastereoselectivity of the enzyme, as shown in Fig. 3. The docking data suggest that Gln-345 might be generally important for chiral recognition by the enzyme.15,16 Use of sucrose phosphorylase in chiral synthesis and chiral resolution strategies for diol-containing acceptor substrates is therefore supported, and this presents a new type of application for enzymatic transglycosylations. Protein engineering that has been employed widely for selectivity enhancement in esterases and lipases should be useful to improve existing (not highly selective) transglycosylation catalysts.17
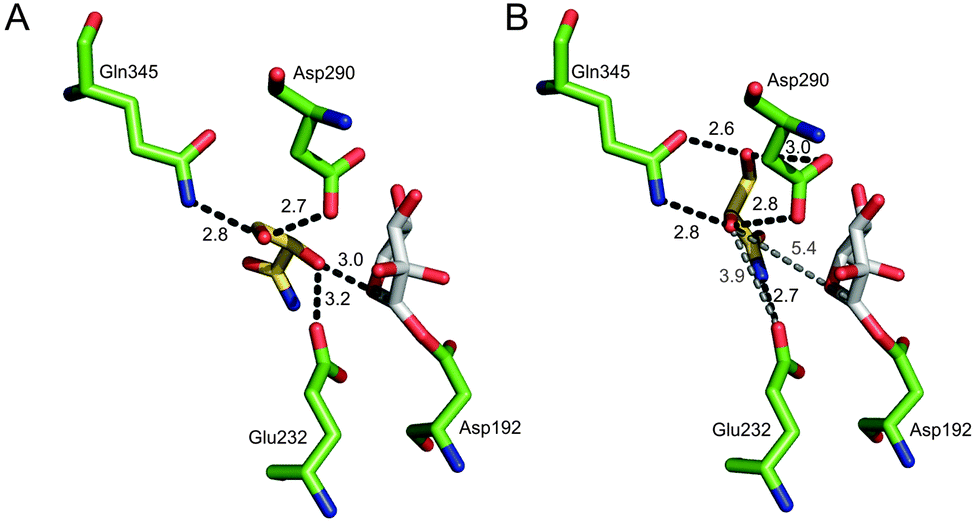 | ||
| Fig. 2 Best-fit docking poses for binding of (R)-2 (A) and (S)-2 (B) to the covalent intermediate of sucrose phosphorylase having a β-glucosyl residue linked to Asp-192 (PDB-entry 2gdv, molecule A). | ||
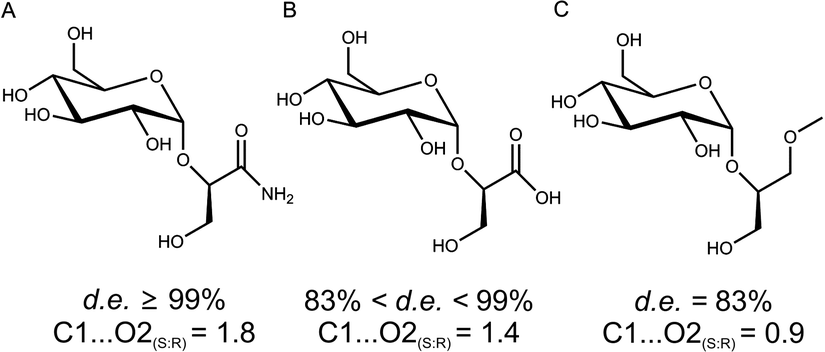 | ||
| Fig. 3 Diastereoselectivity of sucrose phosphorylase in the glucosylation of 1,2-diols correlated with the ratio of reactive atom distances (C1⋯O2) in (S)- and (R)-acceptor poses from molecular docking. | ||
We examined synthesis of 3 as a biomimetic analogue of 4. Reaction optimisation involved mitigation of hydrolysis of 1 at high concentrations of 2 (ESI,† Fig. S4). The glucose released by hydrolysis causes product inhibition and interferes with the reaction by serving as a glucosyl acceptor substrate.6 However, non-productive binding of (S)-2 can also result in inhibition of the enzyme when high levels of racemic 2 are used. Fig. S4 in ESI† shows full time course of synthesis of 3 when 2 was present in equimolar or in 2.5-fold molar excess over 1. Up to two-thirds of 1 were utilised for glucosylation of (R)-2 while the remainder substrate was lost to hydrolysis. Product 3 was obtained in a maximum concentration of 240 mM. Quantitative utilisation of the initial (R)-2 present was possible (ESI,† Fig. S4, panel C). Glucosylation of glucose occurred to a minor extent under these conditions (<5%). Production of 3 was lowered substantially (≤140 mM) upon decreasing the concentration of 2 to 400 mM, now equaling the applied concentration of 1 (ESI,† Fig. S4, panel B), or when using 1 (1000 mM) in excess over 2 (400 mM). Use of 2 in excess over 1 is therefore supported. Procedures for efficient preparative work-up of reaction mixtures from sucrose phosphorylase-catalysed transglycosylations have previously been developed.5,14
Summarising, single-step diastereoselective glucosylation of 2 provides convenient access to the new biomimetic glycoside 3 through chiral synthesis. The same reaction is exploitable through chiral resolution to obtain (S)-2 (ESI,† Fig. S4, panel C). Chiral 1,2-diols are important target molecules in organic chemistry, and their preparation through chiral resolution is therefore of considerable interest.18 Relying on the selectivity of enzymes, biocatalytic glycosylation might present an alternative to reported two-step esterification processes catalysed by lipases/esterases.19
We acknowledge financial support from the Austrian Science Fund FWF (project L586-B03); V. N. Belov (Max Planck Institute for Biophysical Chemistry, Goettingen, Germany) for synthesis of 2; S. Felsinger (Institute of Organic Chemistry, University of Vienna, Austria) for recording several NMR spectra; and C. Araman and N. K. Chu (Institute of Biological Chemistry, University of Vienna, Austria) for help in recording CD spectra.
Notes and references
- P. Bojarova, R. R. Rosencrantz, L. Elling and V. Kren, Chem. Soc. Rev., 2013, 42, 4774–4797 RSC.
- T. Desmet, W. Soetaert, P. Bojarova, V. Kren, L. Dijkhuizen, V. Eastwick-Field and A. Schiller, Chem.–Eur. J., 2012, 18, 10786–10801 CrossRef CAS PubMed.
- J. Kadokawa, Chem. Rev., 2011, 111, 4308–4345 CrossRef CAS PubMed.
- C. A. Weijers, M. C. Franssen and G. M. Visser, Biotechnol. Adv., 2008, 26, 436–456 CrossRef CAS PubMed.
- C. Goedl, T. Sawangwan, M. Mueller, A. Schwarz and B. Nidetzky, Angew. Chem., Int. Ed., 2008, 47, 10086–10089 CrossRef CAS PubMed.
- C. Luley-Goedl, T. Sawangwan, L. Brecker, P. Wildberger and B. Nidetzky, Carbohydr. Res., 2010, 345, 1736–1740 CrossRef CAS PubMed.
- F. van Rantwijk, M. W. V. Oosterom and R. A. Sheldon, J. Mol. Catal. B: Enzym., 1999, 6, 511–532 CrossRef CAS.
- R. Renirie, A. Pukin, B. van Lagen and M. C. R. Franssen, J. Mol. Catal. B: Enzym., 2010, 67, 219–224 CrossRef CAS PubMed.
- G. Lentzen and T. Schwarz, Appl. Microbiol. Biotechnol., 2006, 72, 623–634 CrossRef CAS PubMed (and references therein).
- O. Mirza, L. K. Skov, D. Sprogøe, L. A. van den Broek, G. Beldman, J. S. Kastrup and M. Gajhede, J. Biol. Chem., 2006, 281, 35576–35584 CrossRef CAS PubMed.
- P. F. Frankland, F. M. Whartin and H. Aston, J. Chem. Soc., 1901, 79, 266–274 RSC.
- W. Gaffield, Chem. Ind., 1964, 1460–1461 CAS.
- E. Iizuka and J. T. Yang, Biochemistry, 1964, 3, 1519–1524 CrossRef CAS.
- T. Sawangwan, C. Goedl and B. Nidetzky, Org. Biomol. Chem., 2009, 7, 4267–4270 CAS.
- C. Luley-Goedl and B. Nidetzky, Carbohydr. Res., 2010, 1492–1496 CrossRef CAS PubMed.
- T. Verhaeghe, M. Diricks, D. Aerts, W. Soetaert and T. Desmet, J. Mol. Catal. B: Enzym., 2013, 96, 81–88 CrossRef CAS PubMed.
- U. T. Bornscheuer, Curr. Opin. Biotechnol., 2002, 13, 543–547 CrossRef CAS.
- Y. Zhao, A. W. Mitra, A. H. Hoveyda and M. L. Snapper, Angew. Chem., Int. Ed., 2007, 46, 8471–8474 CrossRef CAS PubMed.
- F. Theil, J. Weidner, S. Ballschuh, A. Kunath and H. Schick, J. Org. Chem., 1994, 59, 388–393 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures used; NMR spectra of 3; the CD spectrum of residual 2 isolated from the reaction mixture; docking poses of R and S-forms of 1,2-diol acceptors glucosylated by sucrose phosphorylase; time course of synthesis of 3; active-site superimposition for sucrose phosphorylases from B. adolescentis and L. mesenteroides; table summarising reaction conditions and product yields. See DOI: 10.1039/c3cc47249c |
| This journal is © The Royal Society of Chemistry 2014 |