 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aerobic oxidation catalysis with stable radicals
Qun
Cao
,
Laura M.
Dornan
,
Luke
Rogan
,
N. Louise
Hughes
and
Mark J.
Muldoon
*
School of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Stranmillis Road, Belfast, UK BT9 5AG. E-mail: m.j.muldoon@qub.ac.uk
First published on 12th March 2014
Abstract
Selective oxidation reactions are challenging when carried out on an industrial scale. Many traditional methods are undesirable from an environmental or safety point of view. There is a need to develop sustainable catalytic approaches that use molecular oxygen as the terminal oxidant. This review will discuss the use of stable radicals (primarily nitroxyl radicals) in aerobic oxidation catalysis. We will discuss the important advances that have occurred in recent years, highlighting the catalytic performance, mechanistic insights and the expanding synthetic utility of these catalytic systems.
 From Left to Right: Laura Dornan, Louise Hughes, Mark Muldoon, Luke Rogan and Qun Cao | Qun obtained a BSc in Chemical Engineering and Technology from the China University of Mining and Technology in 2011. He is carrying out his PhD in the Muldoon group, developing catalytic methods for a number of different oxidation reactions. |
Laura obtained a 1st Class MSci degree in Chemistry from Queen's University Belfast (QUB) in 2012. Her MSci research project was carried out in the Muldoon Group investigating Cu/TEMPO catalysis for the oxidative synthesis of nitriles. She was the recipient of a Nuffield Undergraduate Research Bursary in 2011 and in 2013 she carried out an internship at Genentech in San Francisco. She is carrying out her PhD in the Muldoon Group exploring new methods for selective oxidation catalysis. |
Luke obtained a 1st Class BSc degree in Chemistry with Extended Studies in Europe from QUB in 2012. During his undergraduate studies he spent a year at the University of Zaragoza in Spain, where he carried out research in synthetic organometallic chemistry. He joined the Muldoon Group in September 2012 and his PhD studies are investigating aqueous oxidation reactions and the use of solid supported catalysts. |
Louise obtained a 1st Class MSci degree in Chemistry from QUB in 2013. Her MSci research project was carried out in the Muldoon Group investigating the use of sterically unhindered nitroxyl radicals for aerobic oxidation catalysis. She commenced her PhD in the Muldoon Group in September 2013. |
Mark obtained his BSc from the University of Strathclyde in Glasgow in 1998. He then worked as a process scientist for Boots PLC before returning to the University of Strathclyde for his PhD (1999–2003). He carried out postdoctoral research at the University of Notre Dame, Indiana, USA (2003–2005). From 2005–2007 he held a Ramsay Memorial Fellowship at the University of St Andrews. In 2007 he was appointed to a Lectureship in Inorganic Chemistry at QUB. His research interests are focused on catalysis, (particularly selective oxidation reactions), neoteric solvents and reaction engineering. |
Introduction
Developing practical and sustainable catalytic methods for selective oxidation reactions is an important challenge. In the production of fine chemicals and pharmaceuticals, oxidations are often problematic. For example, in a study by leading pharmaceutical companies which examined the reactions commonly used in the synthesis of drug candidates, it was found that oxidations were often avoided.1 In their conclusions, the authors stated: “In contrast to reductions, there are relatively few atom efficient, chemoselective and environmentally acceptable oxidation methods. As a consequence, oxidations are often designed out of syntheses. The discovery of new chemoselective oxidations, particularly if catalytic, would greatly increase flexibility in synthetic design”.1In terms of sustainability, O2 is the ideal terminal oxidant. It is readily available and H2O is usually the by-product of catalytic oxidation reactions. The importance of developing better aerobic oxidation catalysts means there is now an increasing emphasis on this challenge and it is becoming a fast moving research area. A plethora of metal catalysts (both homogeneous and heterogeneous) have been reported for a wide range of oxidation reactions; ranging from alcohol oxidation to C–H bond functionalisation.2
In this review we will concentrate on the use of stable radical catalyst systems. We believe these catalysts have attributes that make them viable for larger scale applications. It is common strategy that these stable radicals are combined with metal co-catalysts and these are normally first row transition metals such as copper and iron. Although platinum group metals such as Pd, Pt, Ru and Ir have a long history in oxidation catalysis, these metals are expensive and the cost of such metals is only likely to get higher. Recently the British Geological Society published a report on the “supply risk” for chemical elements,3 and platinum group metals were identified as high risk elements. In contrast, copper and iron were rated as very low risk. Platinum group metals are also substantially more toxic than copper and iron and their presence in pharmaceutical and dietary products must be kept to very low levels.4 Platinum group metals are “Class 1 metals: metals of significant safety concern”, while copper comes under “Class 2 metals: metals of low safety concern” and iron is under “Class 3 metals: metals of minimal safety concern”.4 Furthermore, catalysts based on copper and iron tend to have better tolerance of other functional groups and heteroatoms, unlike noble metal catalyst systems, which we will highlight later. This is an important trait when oxidations are required on poly-functionalised molecules. We will also give examples of stable radical catalysts systems that do not employ any transition metal co-catalysts.
In 2010, Ciriminna and Pagliaro reviewed some of the recent industrial oxidation processes that utilise the stable radical 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) and its derivatives.5 In this review, only one of the industrial examples was aerobic and the rest all utilised Anelli–Montanari6 type conditions. In this method the oxoammonium salt of TEMPO carries out alcohol oxidation and hypochlorite is added in excess (Fig. 1). Although this approach can be very effective, hypochlorite (i.e. bleach) is not compatible with certain chemicals, for example ammonia (see nitrile synthesis later). An additional problem is that this method can result in unwanted chlorinated by-products.7 Consequently, there are some important drivers for moving towards halide free systems which use O2 as the terminal oxidant.
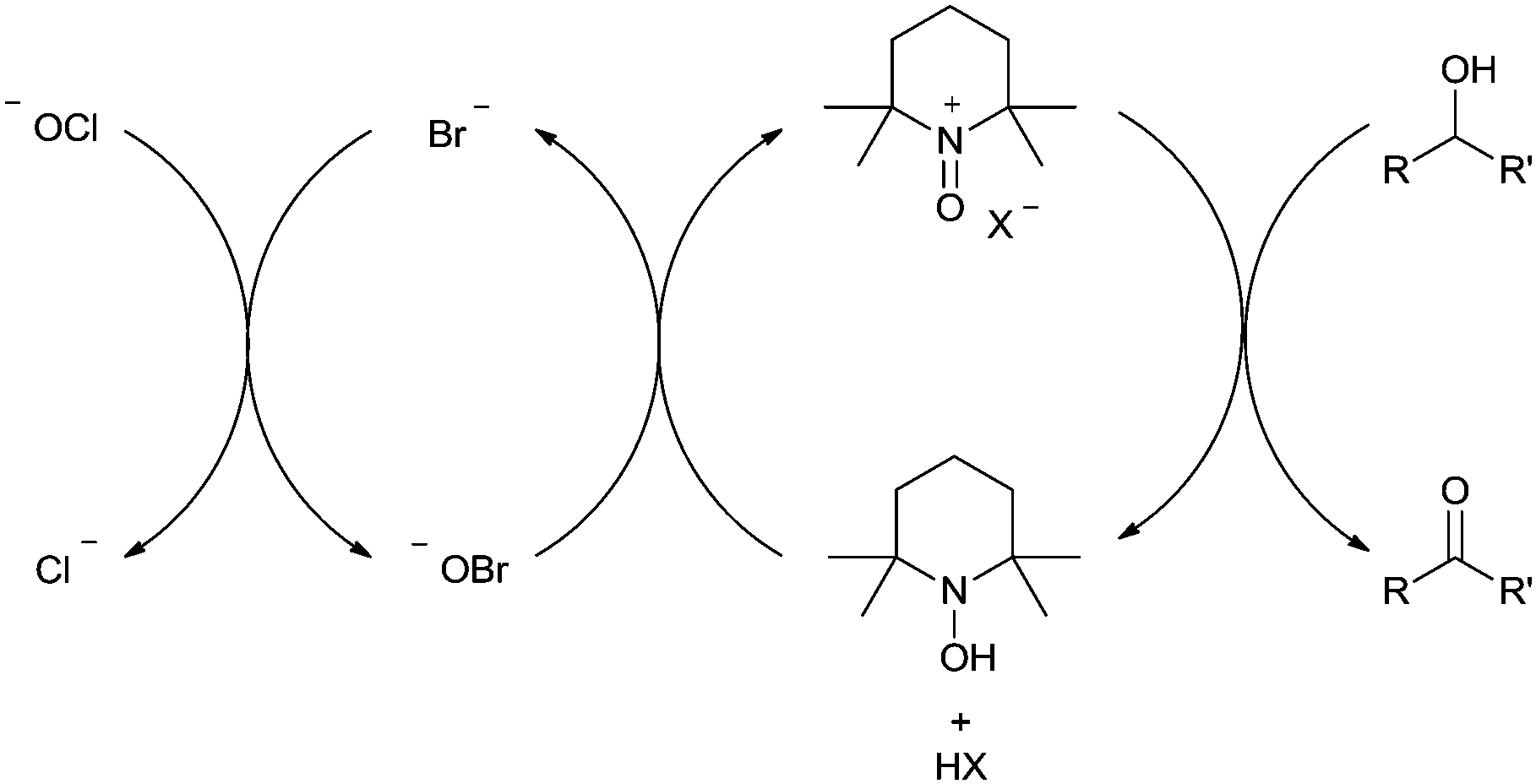 | ||
| Fig. 1 Anelli–Montanari protocol where oxidation of the alcohol takes place via the oxoammonium salt. | ||
Scope of the article
This review will focus on recent advances in aerobic oxidations using stable radicals as catalysts for applications in synthetic chemistry. In most cases the radicals in question will be nitroxyl radicals; however there are a few examples that involve other classes of stable radical. It is worth mentioning some preceding manuscripts in this area. In 2004 and 2006 Sheldon and Arends published reviews on the use of nitroxyl radicals for catalytic oxidations.8 In 2009, Bobbitt et al. reviewed the use of nitroxides and oxoammonium salts for the catalytic oxidation of alcohols.9 In 2011, Tebben and Studer published a review on the use of nitroxide radicals for a range of applications in synthesis and in polymer chemistry.10 Wertz and Studer recently published a review on transition-metal-free nitroxide-catalysed systems which covers both stable and non-persistent radicals.11 Very recently, Allen et al. published an all-encompassing review on copper catalysed aerobic oxidations (discussing the literature up to 2011), and this examined a significant number of copper/TEMPO studies.2h A book published by Hicks (ed.) discusses in detail many aspects of stable radicals and their chemistry.12We are focused on stable radicals and as such will not discuss the use of N-hydroxyphthalimide (NHPI) which is used to generate the nitroxyl radical phthalimide-N-oxyl (PINO). The PINO radical (and similar analogues) have been utilised for a range of aerobic oxidation reactions. PINO has been shown to be more reactive than TEMPO, however, it is not stable and is usually formed from NHPI in situ by means of an initiator. Consequently this falls outside the remit of this article; however it has been reviewed elsewhere13 and is discussed in some of the reviews already mentioned.8,10,11
The use of stable radicals in catalysis is an area that has gathered momentum in the last few years and as is appropriate in such a short review, we will concentrate on recent developments to avoid repetition of the discussions in these excellent earlier articles. Alcohol oxidation has received the most attention to date and we will therefore cover this important fundamental reaction in more detail, also outlining the latest mechanistic insights. Stable radical catalysts are increasingly being utilised for a variety of different oxidation reactions and we will highlight some of the new applications that are emerging.
Unhindered nitroxyl radicals
At this point it is worth introducing unhindered nitroxyl radicals as these will be discussed in later sections. The majority of studies have focused on the use of TEMPO, which is undoubtedly the most well-known and well-studied stable radical. Recently the use of sterically unhindered nitroxyl radicals has emerged. Such radicals have been known since the 1960s14 but until recently they have not been exploited for oxidation catalysis. Unlike TEMPO, unhindered radicals such as those shown in Fig. 2 possess α-hydrogens but are still stable, as they are protected from disproportionation (to the hydroxylamine and a nitrone), because the resulting double bond would form at a bridgehead, violating Bredt's rule.15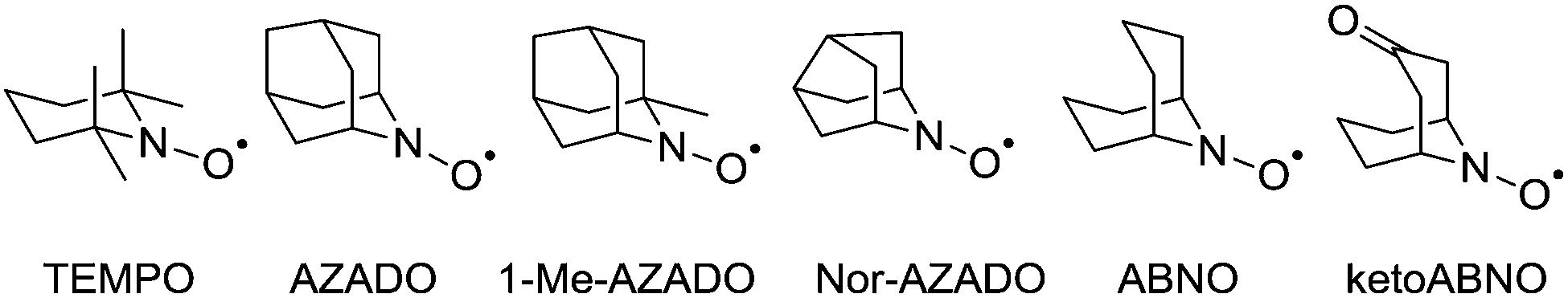 | ||
| Fig. 2 Structural comparison of nitroxyl radicals. From left to right: 2,2,6,6-tetramethylpiperidinyloxy (TEMPO), 2-azaadamantane N-oxyl (AZADO), 1-methyl-2-azaadamantane N-oxyl (1-Me-AZADO), 9-azanoradamantane N-oxyl (Nor-AZADO), 9-azabicyclo[3.3.1]nonane N-oxyl (ABNO), 9-azabicyclo[3.3.1]nonan-3-one N-oxyl (ketoABNO). | ||
These types of radicals were initially examined in fundamental physical-organic studies, and it has been known for some time that ABNO is more reactive than TEMPO.16 However, only recently have these radicals been used for oxidation chemistry in the same manner as TEMPO. Early oxidation studies used electrochemical methods,17 trichloroisocyanuric acid18 or bleach19 as the oxidant to generate the oxoammonium salt. It was shown that such unhindered radicals could carry out oxidations faster than TEMPO and, perhaps, more importantly, they could oxidise sterically hindered alcohols that TEMPO could not. Fig. 3 illustrates the ability of ABNO to efficiently oxidise a bulky alcohol compared to TEMPO (and 1-Me-AZADO) using Anelli–Montanari conditions.
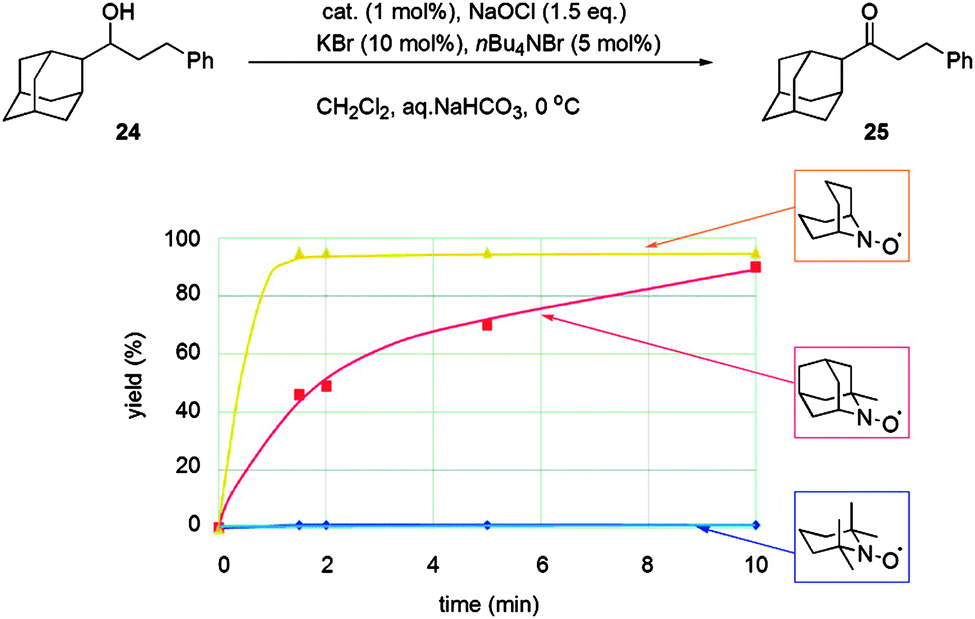 | ||
| Fig. 3 Illustration of how steric hindrance between radicals and substrates affects reactivity using the Anelli–Montanari protocol. Reproduced with permission from ref. 19b © 2009 American Chemical Society. | ||
The use of these stable radicals for aerobic oxidation catalysis has now been reported for a number of different applications and we will discuss these in the relevant sections below.
Alcohol oxidation
Alcohol oxidation is a fundamental reaction in organic chemistry, and while traditional methods may be trivial to use on a small scale, this reaction poses a real challenge when it has to be carried out on an industrial scale.1 There has therefore been a considerable amount of effort put into developing sustainable aerobic catalytic methods for this reaction. We believe that the use of stable radicals for aerobic alcohol oxidation has emerged as a very promising approach. Nitroxyl radicals are often combined with first-row transition metal co-catalysts, although the use of nitrites or nitric acid to produce NOx, which then acts as a co-catalyst, is also a method that is showing great promise.Copper
The first example of aerobic alcohol oxidation using copper and a nitroxyl radical was reported in 1966.20 Brackman and Gaasbeek combined di-tert-butylnitroxyl with a copper(II) phenanthroline complex for the oxidation of methanol. However, the most well-known and widely studied system employs copper complexes combined with TEMPO, which was first reported by Semmelhack and co-workers in 1984.21 Since that report, numerous studies have explored this catalyst system, investigating ligands,22 neoteric solvent systems,23 immobilisation,24 reaction engineering25 and oxidation of renewable substrates.26 However, we wish to focus on the recent reports, primarily from the Stahl Group, which exemplify the potential of Cu/nitroxyl catalysis for synthesis and also give further mechanistic insights. Prior to discussing these papers we want to highlight some seminal studies that shaped the development of this catalyst system.The original Semmelhack study combined TEMPO with CuCl using DMF as the solvent and O2 bubbling through the system.21 This catalyst system could oxidise 1° benzylic and allylic alcohols at 25 °C, but was not effective for aliphatic alcohols. It was shown to preferentially oxidise 1° over 2° alcohols. Later, Sheldon and co-workers developed a system which utilised Cu(II) salts, 2,2′-bipyridine (bpy) as a ligand and t-BuOK as a base in a CH3CN/H2O (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent mixture.27 This catalyst system was able to carry out oxidations at 25 °C using ambient air as the source of O2. For activated alcohols, high yields of product were obtained in 2–5 hours using 5 mol% catalyst loadings. In order to obtain a high yield for an unactivated primary alcohol (1-octanol) the reaction required heating at 40 °C for 24 hours with a higher loading of TEMPO (7.5 mol%). This catalyst was also found to be selective for primary alcohols. Kinetic studies27b,c led the authors to suggest that the reaction mechanism may have similarities to that of the copper protein galactose oxidase,28 with TEMPO playing the role of the protein-bound phenoxyl radical. This suggestion of a copper-centred dehydrogenation differed significantly from the ‘oxoammonium’ type mechanism previously proposed by Semmelhack.21
:1) solvent mixture.27 This catalyst system was able to carry out oxidations at 25 °C using ambient air as the source of O2. For activated alcohols, high yields of product were obtained in 2–5 hours using 5 mol% catalyst loadings. In order to obtain a high yield for an unactivated primary alcohol (1-octanol) the reaction required heating at 40 °C for 24 hours with a higher loading of TEMPO (7.5 mol%). This catalyst was also found to be selective for primary alcohols. Kinetic studies27b,c led the authors to suggest that the reaction mechanism may have similarities to that of the copper protein galactose oxidase,28 with TEMPO playing the role of the protein-bound phenoxyl radical. This suggestion of a copper-centred dehydrogenation differed significantly from the ‘oxoammonium’ type mechanism previously proposed by Semmelhack.21
The catalyst system was further improved by Kumpulainen and Koskinen.29 They carried out kinetic studies, investigating the effects of the different components in the system. Bpy was used as the ligand along with Cu(II) salts to explore the effect of the base in detail. It was found that better performance could be obtained using organic bases such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and N-methylimidazole (NMI). They also found that pure acetonitrile gave the best results (as opposed to an aqueous mixture). Kinetic studies found a first-order (1.15) kinetic correlation for TEMPO and a second-order correlation (2.25) for copper, which supported the idea that a binuclear copper intermediate was formed during the reaction. The optimised system demonstrated improved reactivity and could oxidise aliphatic alcohols in good yields at room temperature. Utilising 3 mol% Cu(OTf)2, bpy, TEMPO, NMI and DBU, 1-decanol could be oxidised to almost complete conversion in 5 hours. With the addition of 3 Å molecular sieves to remove traces of water, complete conversion was achieved in 3 hours with 95% isolated yield. These reactions were carried out under an atmosphere of O2 (balloon), although during their kinetic studies (which focused on trans-2-hexen-1-ol as a model substrate) they showed that the use of air only decreased the rate slightly. This more reactive system could oxidise secondary aliphatic alcohols, albeit at a higher temperature (40 °C) and longer reactions times (59% conversion in 48 hours).
In 2011, Hoover and Stahl reported that CuI salts were superior to CuII salts in a system which used NMI as a base.30 It can be seen in Fig. 4 that Cu(I) salts in conjunction with bpy and NMI in pure acetonitrile resulted in improved reactivity.
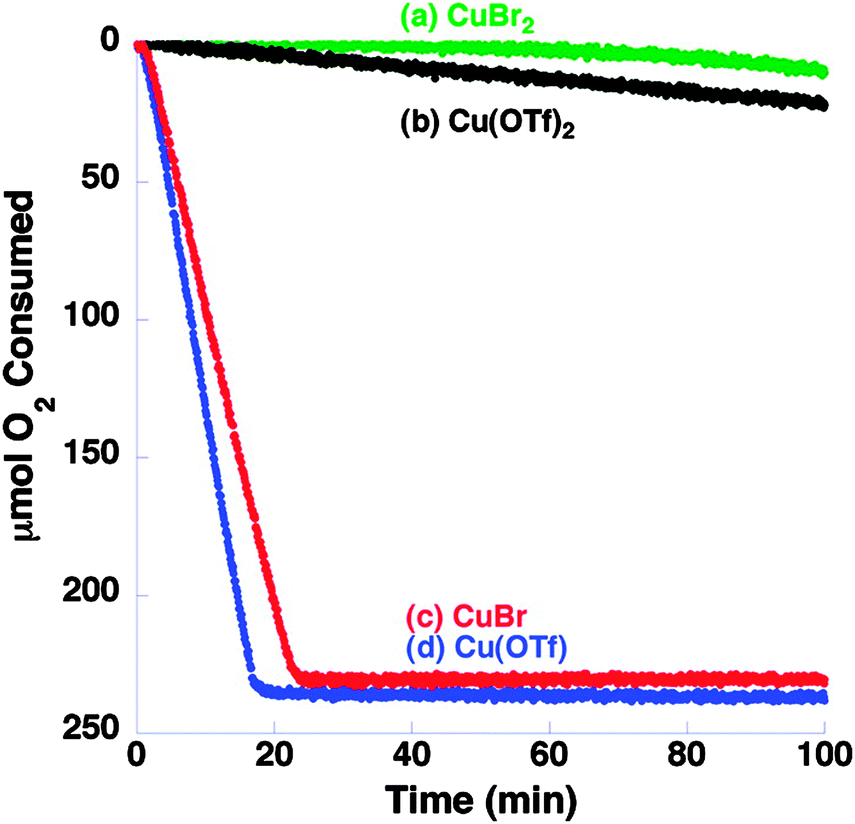 | ||
| Fig. 4 Influence of Cu source on the oxidation of benzyl alcohol oxidation at 27 °C in CH3CN using Cu source (5 mol%), bpy (5 mol%), TEMPO (5 mol%) and NMI (10 mol%). Reproduced with permission from ref. 30 © 2011 American Chemical Society. | ||
This study also demonstrated that Cu/TEMPO can oxidise a wide range of substrates and is tolerant of many functional groups. Fig. 5 shows a sample of the substrate scope for this system. As part of their studies they examined how the Cu(I)/TEMPO system compared to other homogeneous oxidation catalysts. Pd(OAc)2/pyridine and RuCl2(PPh3)3/TEMPO systems were tested, which showed that these precious metal catalysts struggled to oxidise substrates containing alkene, alkyne, ether, sulphur and nitrogen functionalities.
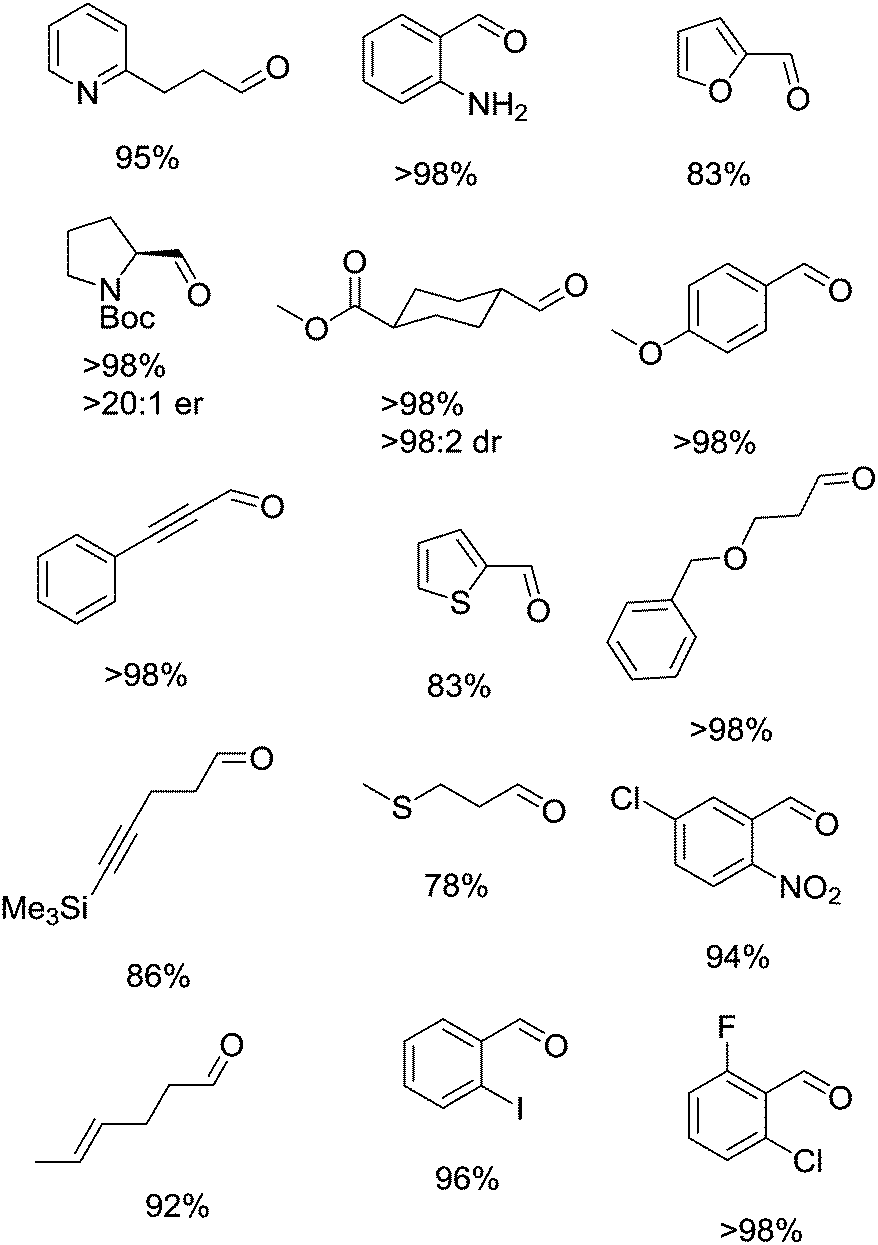 | ||
| Fig. 5 Examples of the carbonyl products that can be produced using (bpy)CuI/TEMPO (5 mol%) and NMI (10 mol%) in CH3CN. | ||
As we have mentioned, Cu/TEMPO systems are often selective for primary alcohols over secondary alcohols. This study also looked at the chemoselectivity using a number of different diol substrates, studying both steric and electronic effects (i.e. benzylic vs. aliphatic). It was shown that it is possible to obtain selectivity for 1° over 2° for a variety of alcohols. In some cases, the (bpy)Cu(OTf)/TEMPO/NMI system was too reactive and improved selectivity was obtained by varying the choice of copper salt or base. For example, in the case of 1° benzylic vs. 2° benzylic substrates, Cu(I) salts were too active and also oxidised the 2° alcohol group to the ketone. Using the less reactive CuBr2 allowed selective oxidation of only the 1° alcohol. In other cases, switching from Cu(OTf) to CuBr was sufficient to improve the selectivity.
They also discussed the limitations of the catalyst, highlighting a number of problematic substrates. It was found that homobenzylic substrates were over-oxidised with oxygenation occurring at the benzylic position. Terminal alkynes were found to produce a mixture of unwanted products. Substrates with the ability to chelate such as vicinal diols or those with adjacent heteroatoms (e.g. 2-pyridinemethanol) led to very low conversions. Substrates with a phenol substituent were also shown to be unreactive. In these cases it would seem that the substrates bind to the Cu and form unreactive complexes.
In most cases, substrates could be oxidised at room temperature using ambient air (“open flask”). This makes this method31 very safe and accessible to those wanting to use it on a small scale; indeed it has even been shown to be suitable as an undergraduate laboratory experiment.32 On a larger scale, this “open flask” approach is obviously not practical. In industry it is common practice to use dilute oxygen mixtures (e.g. <10% O2 in N2) so that the gas mixture is essentially non-flammable. The most scalable approach would be to operate under continuous flow conditions. Stahl and co-workers have recently published a report demonstrating the use of Cu(I)/TEMPO for alcohol oxidation under continuous flow conditions.33 In this case the system used 35 bar of 9% O2 in N2 and they heated a tubular reactor to temperatures up to 100 °C. High yields of aliphatic aldehydes could be obtained with residence times from 30–45 min, while activated alcohols only required residence times of ≤5 min. In the case of benzyl alcohol to benzaldehyde they demonstrated the reaction on a larger scale to produce 100 g of product.
The initial Cu(I)/TEMPO studies by Stahl and co-workers focused on the substrate scope and methods. This was followed up in 2013 with mechanistic studies for this catalyst system.34,35 These studies are the most comprehensive mechanistic studies to date with a range of techniques employed to help propose a mechanism. Gas chromatography, gas-uptake, in situ IR, cyclic voltammetry, EPR and UV-visible spectroscopy were all utilised and detailed studies of the kinetic isotope effect were carried out. The catalytic cycle they proposed is shown in Fig. 6.
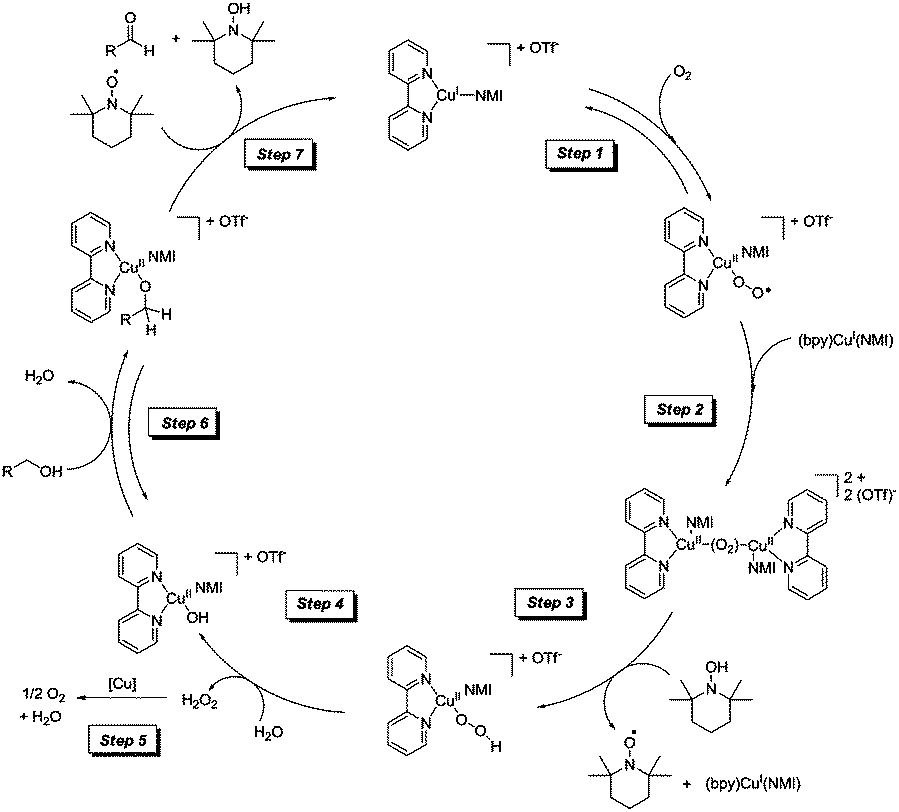 | ||
| Fig. 6 Catalytic cycle for CuI/TEMPO aerobic oxidation of alcohols.34 | ||
Both activated (benzyl alcohol) and unactivated (cyclohexylmethanol) substrates were examined and it was found that these different classes of substrate had different turnover-limiting steps. In the case of benzyl alcohol (which is easier to oxidise) they found that oxidation of the catalyst by O2 was the limiting step. As can be seen in Fig. 6, catalyst oxidation is proposed to occur via a binuclear Cu2O2 intermediate. In the case of the unactivated substrate, cyclohexylmethanol, it was found that both substrate oxidation and catalyst oxidation affected the rate. It is a general phenomenon that aliphatic alcohols are more difficult to oxidise and many catalysts struggle with such substrates, even under more forcing conditions. In the case of this Cu/TEMPO system the pKa of aliphatic alcohols means that the formation of Cu-alkoxide species will form less readily and the bond strength of the α-C–H bond is significantly greater for aliphatic alcohols; which will clearly affect the product forming step(s). In this mechanism it is proposed that hydrogen atom abstraction (from the α-C–H) occurs via a bimolecular reaction between the Cu-alkoxide and TEMPO. In this study they found no evidence of a Cu–TEMPO adduct being involved in this step; which is in agreement with another recent study by Bonnet and co-workers.36 As we mentioned earlier, oxoammonium salts have been proposed as active species in Cu systems, however in this Cu(I)/TEMPO study they utilised cyclic voltammetry and EPR to show that Cu(II) could not oxidise TEMPO to its oxoammonium (TEMPO+) ion under the reaction conditions.
As outlined earlier, there are a number of closely related Cu/TEMPO systems (i.e. Semmelhack, Sheldon, Koskinen and now Stahl). In the second mechanistic report by the Stahl group,35 they examined the differences between these systems. The influence of ligand, solvent, base and Cu source were studied, giving further mechanistic insights into the effect of these components. Worth highlighting here is the reason for the rate acceleration found when CuI (rather than CuII) salts are employed with NMI as the base. It is believed that the in situ formation of a hydroxide base (step 4 in Fig. 6) allows the CuII-alkoxide species to be readily formed. In this system, it appears as though NMI may be acting as a ligand. If a CuII source is used, such a Cu hydroxide species is not formed and it is necessary to add a stronger base (e.g. DBU) in order to obtain a fast reaction. CuIOTf/NMI and CuII(OTf)2/DBU may have similar efficiencies, however the authors point out that using a system with a weaker base (i.e. NMI) can be very useful in some cases, as strong bases can lead to epimerization of stereocentres.
Although we have only highlighted these recent studies, there have been a variety of mechanisms and variations of mechanisms suggested for Cu/TEMPO catalysed alcohol oxidation.27,29,37–40 Of course, it is possible that a number of variations are feasible and different pathways will depend on the reaction components and conditions. Nonetheless, such mechanistic studies are extremely valuable as the insights they provide can lead to the design of better catalysts. Indeed, very recently copper based systems were further improved by utilising such mechanistic understanding. As already discussed, the Cu/TEMPO systems allow primary alcohols to be selectively oxidised in the presence of secondary alcohols. While this may be a significant advantage in some cases, the oxidation of secondary alcohols is often desired and Cu/TEMPO is not a suitable catalyst. One of the reasons for the poor performance of secondary alcohols is believed to be due to steric hindrance (as secondary alcohols are bulkier) and the final step in the mechanism involves a bimolecular reaction between the Cu-alkoxide and TEMPO. It was recently shown by Steves and Stahl that this can be addressed by switching from TEMPO to the sterically unhindered nitroxyl radical ABNO.41 The optimised system consisted of 5 mol% Cu(MeCN)4OTf, 5 mol% 4,4′-dimethoxy-2,2′-bipyridine (MeObpy), 10 mol% NMI and 1 mol% ABNO (Fig. 7).
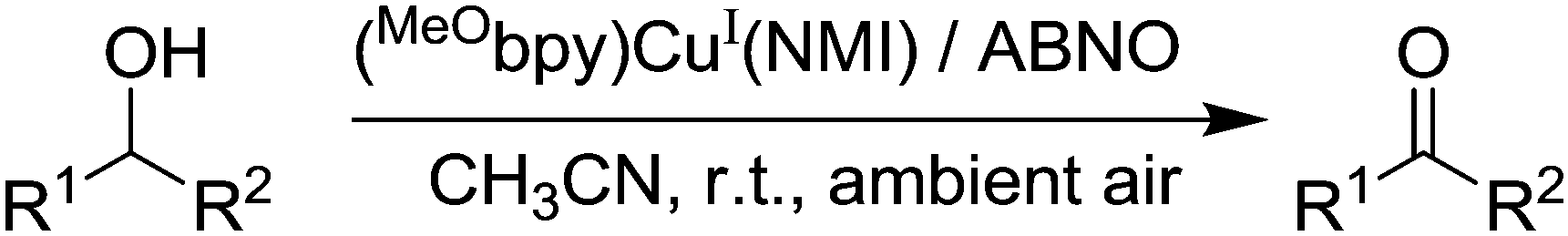 | ||
| Fig. 7 CuI/ABNO aerobic oxidation of alcohols. | ||
The Cu(I)/ABNO system can effectively oxidise a wide range of secondary alcohols, including substrates with bulky groups close to the alcohol group, such as 2,2-dimethyl-3-octanol, menthol and 2,2-dimethyl-1-phenyl-1-propanol. Using the conditions described, high isolated yields of product could be obtained in just 1 hour in most cases. As might be expected, the catalyst can still oxidise primary alcohols and has a similar functional group tolerance to that of Cu/TEMPO, with similar limitations also (e.g. terminal alkynes, phenols and primary homobenzylic alcohols).
Although the Steves and Stahl study41 included some amino functionalised substrates, very recently Iwabuchi and co-workers reported the use of Cu/AZADO for the oxidation of a wide range of unprotected amino alcohols (Fig. 8).42 In initial optimisation experiments they found that a number of unhindered radicals were far superior to TEMPO. From these studies the Cu/AZADO system was chosen and explored for the oxidation of a wide range of substrates, with AZADO loadings of 1–5 mol% used. It was demonstrated that along with tertiary amines, substrates containing primary and secondary alcohols could also be oxidised in good to excellent yields. They compared the AZADO catalyst system to traditional oxidation methods (pyridinium chlorochromate, Swern, Dess–Martin periodinane and tetrapropylammonium perruthenate) for a number of model substrates and found that the AZADO system could deliver better yields. Furthermore, they synthesised two natural products (myosmine and (−)-mesembrine), which are prepared by the oxidation of amino alcohol precursors. In these cases, higher catalyst loadings were required (10 and 15 mol% respectively), however it does highlight the potential of these catalyst systems.
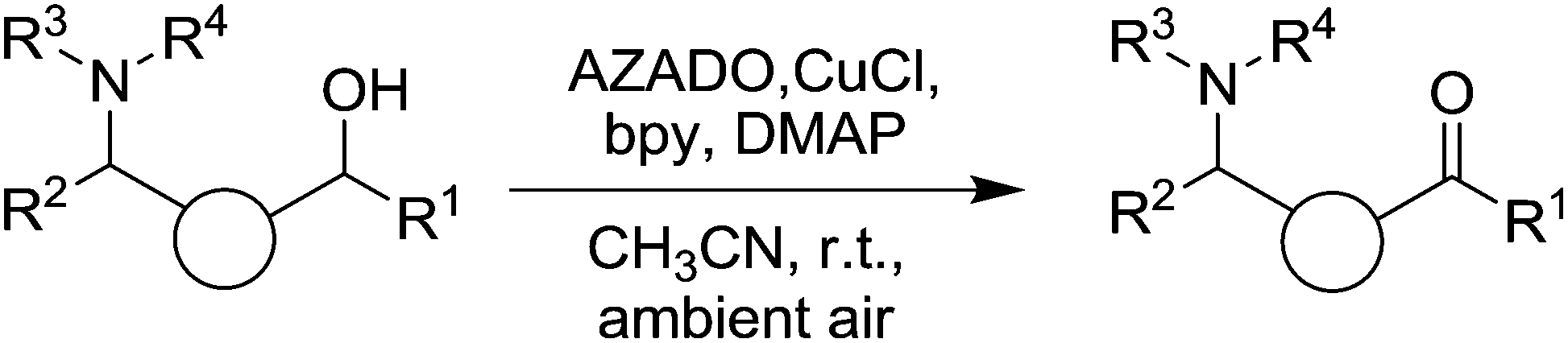 | ||
| Fig. 8 CuI/AZADO aerobic oxidation of amino alcohols. | ||
Iron
A number of publications have reported aerobic Fe/TEMPO systems for catalytic alcohol oxidation. The exact nature of the catalyst often varies and an assortment of iron salts and TEMPO derivatives have been reported;43–47 but there are some general trends. Iron(III) salts are normally employed and unlike Cu/TEMPO systems, Fe/TEMPO catalysts don't usually require any base or organic ligands. The best performance also appears to be obtained from weakly coordinating solvents (e.g. dichloroethane) unlike Cu/TEMPO which normally uses acetonitrile. The substrate scope for these catalysts is also very good and can tolerate alkenes, alkynes and heteroatoms. It is fair to say that Fe/TEMPO systems have not yet been subjected to detailed mechanistic studies, so the exact mechanism(s) is still open to conjecture. Recent studies with MCl3(η1-TEMPO) (M = Fe, Al) showed that these complexes can oxidise alcohols,48 although the majority of catalytic studies utilise Fe(NO3)3 as the iron salt or use NaNO2 as an additive. It is therefore likely that NOx is playing a key role in these systems and this has been suggested by some authors.43,47 It has been known for some time that NO2 can oxidise nitroxyl radicals such as TEMPO to their oxoammonium salts (Fig. 9),49 so this is certainly a very plausible explanation for the use of nitrate salts or nitrite additives. It has also been suggested that NO2 could also assist in the re-oxidation of Fe(II) to Fe(III).47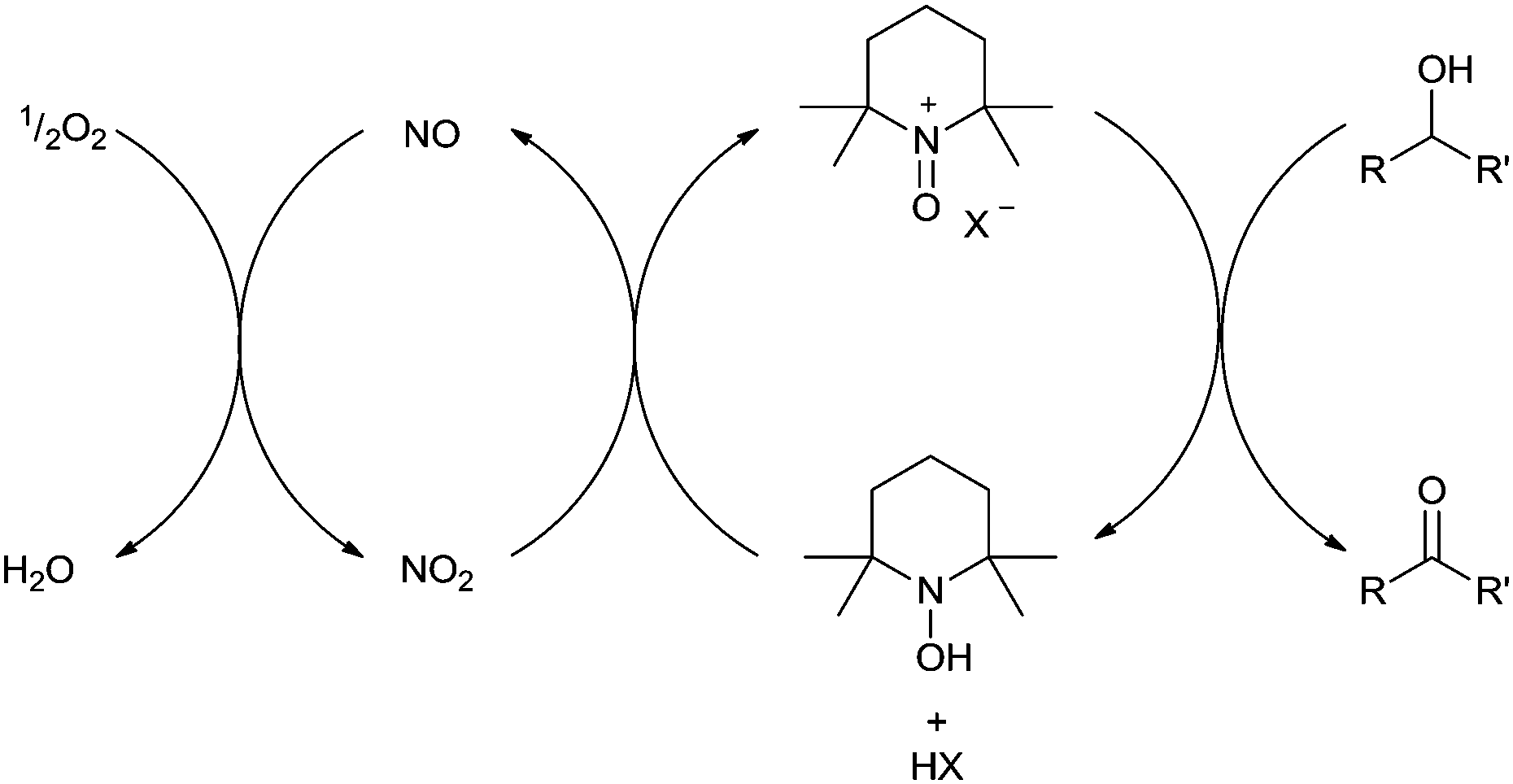 | ||
| Fig. 9 Aerobic oxidation of alcohols via the oxoammonium cation coupled with NOx. | ||
Unlike the Cu/TEMPO system, Fe/TEMPO readily oxidises secondary alcohols. This could be because the oxoammonium salt is the active oxidant, although arguably even if iron/TEMPO did work in unison, one might not expect the same preference for primary alcohols. Fe/TEMPO systems do not require an additional ligand (such as bpy) and therefore they are sterically less hindered compared to Cu/TEMPO systems.
We will now highlight some of the more recent Fe/TEMPO papers. In 2011 Ma et al. improved the performance and substrate scope of previous Fe–TEMPO catalyst systems, by using NaCl as an additive.47 The optimised components for room temperature alcohol oxidation were Fe(NO3)3·9H2O, TEMPO and NaCl using an oxygen balloon with 1,2-dichloroethane (DCE) as solvent. Some examples of the carbonyls that were prepared are shown in Fig. 10.
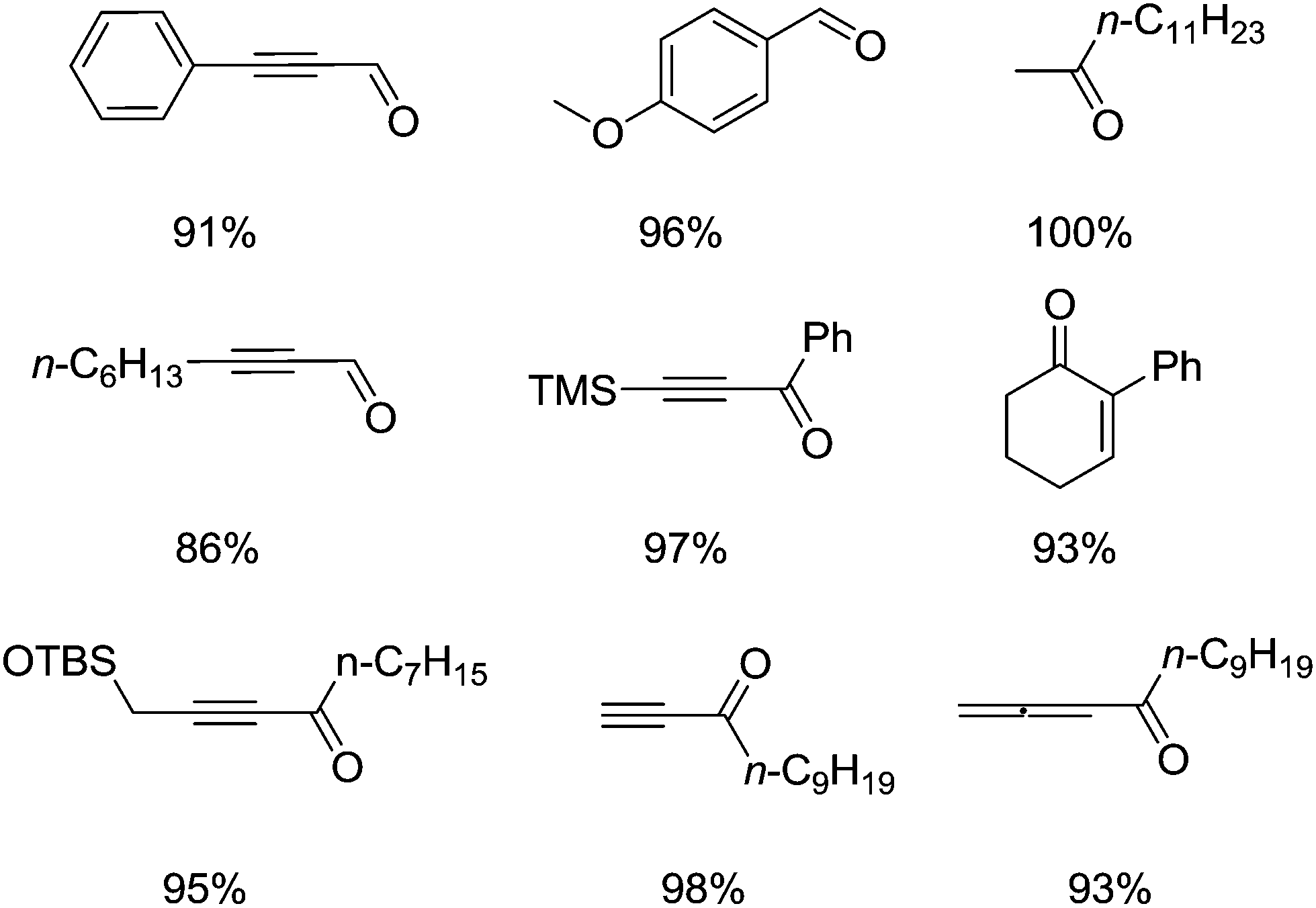 | ||
| Fig. 10 Some examples of products produced by the Fe(NO3)3·9H2O/TEMPO/NaCl system. | ||
This Fe/TEMPO system readily oxidised standard model alcohols such as primary and secondary allylic, benzylic and unactivated aliphatic alcohols. A range of primary and secondary allenols were also oxidised without over-oxidation. The catalyst was tolerant of functional groups such as silyl ethers. Primary and secondary propargylic alcohols, including substrates with a terminal alkyne group, were oxidised. Fe/TEMPO systems are also tolerant of heteroatoms such as oxygen, sulfur and nitrogen. It is worth noting that in the case of nitrogen normally only pyridine type substrates have been shown in publications to date. We suspect that some amines may not be compatible due to the presence of NOx. For example, anilines would likely lead to the formation of azo compounds (see later for such an example in a NOx type system).
Using 1-phenylethanol as a model substrate this research group have demonstrated the reaction on a larger scale (4 mol) and also with air as the oxidant. Subsequently, they applied the Fe(NO3)3·9H2O/TEMPO/NaCl/DCE catalyst system to the oxidation of indole-carbinols,50 a range of propargyl alcohols,51 and allylic alcohols.52 In the latter case, they found that it was possible to produce the α,β-unsaturated enals and enones with retention of the C–C double-bond configuration.
Gao and co-workers recently reported two Fe/TEMPO catalyst system that utilise silica. Their first report was a rare example that did not use Fe(NO3)3 or NaNO2; instead silica gel was coupled with FeCl3·6H2O/TEMPO.53 It was thought that the silica gel acted as a support for the catalyst. The optimised catalyst system used toluene as the solvent and a temperature of 80 °C. Reactions were carried out in a pressurised reactor and utilised either 5 bar of O2 or air, with air requiring longer reaction times. The catalyst did not perform well for unactivated aliphatic alcohols. They then followed this study with a different silica based system which showed improved reactivity.54 In this case TEMPO was covalently bound to a silica support and combined with FeCl3·6H2O/NaNO2. The heterogeneously supported TEMPO was prepared via reductive amination of 4-oxo-TEMPO with amine-functionalised SBA-15 (an approach to immobilise TEMPO that has previously been reported by Bolm and co-workers55). Using benzyl alcohol as a model substrate the authors could achieve very efficient oxidation to benzaldehyde with a loading of just 0.01 mol% of the heterogeneous radical, with 8 mol% FeCl3·6H2O and 10 mol% NaNO2, in toluene and O2 (1 atm) at 25 °C. TEMPO is clearly the most expensive part of these catalyst systems and in this study they not only showed that reduced loadings of TEMPO could be used but that the catalyst could also be recycled. For benzyl alcohol oxidation both the iron salt and heterogeneous TEMPO were recycled together (5 runs). They also showed that SBA-15-TEMPO could be recycled on its own (i.e. with fresh FeCl3·6H2O and NaNO2) and used for 10 reactions without significant loss in activity or selectivity for the benzaldehyde product. The oxidation of a range of substrates was shown with loadings of SBA-15-TEMPO from 0.1 to 1 mol%. This catalyst system had enhanced performance for unactivated substrates, although primary aliphatic alcohols were not converted as readily as secondary aliphatic alcohols. The authors also demonstrated for a series of alcohols that atmospheric air could be employed, albeit with longer reactions times.
Obermayer et al. described a continuous flow approach employing a microreactor.56 In this case, the Fe–TEMPO system comprised of a heterogeneous iron oxide nanoparticle catalyst stabilised on a mesoporous aluminosilicate support, with TEMPO dissolved in solution and flowed through the reactor. The oxidation of benzyl alcohol to benzaldehyde was used as a model reaction. It was found that the activity of the Fe/Al-SBA-15 nanoparticle catalyst (1 wt% Fe) was enhanced by the addition of TEMPO. They could achieve a yield of 42% benzaldehyde in a single pass of the reactor, however compared to many of the systems we have discussed, the reaction conditions were rather harsh; 120 °C and 35 bar of pure oxygen. Nonetheless, this illustrates an important feature of using continuous flow microreactor systems; they enable reaction conditions to be employed that would otherwise be very unsafe. Indeed, we believe that such reaction engineering solutions will be central for aerobic reactions to be used industrially.
Cobalt and manganese
The use of cobalt and manganese co-catalysts with TEMPO and its derivatives has been known for some time.57–61 These studies have utilised nitrate salts, often employing the combination of both Co and Mn salts and using acetic acid as the solvent. The conditions would suggest that in these reactions the substrates are oxidised by the oxoammonium salt. Although there have not been extensive substrate scope studies, these catalyst systems can oxidise both secondary and primary alcohols and can convert unactivated aliphatic substrates; although secondary aliphatic alcohols require longer reaction times than their primary counterparts. There have also been reports with heterogeneous Cu/Mn oxide co-catalysts which do not employ acidic solvents.62–64 In these cases, low loadings of TEMPO were used and the heterogeneous catalyst was recycled, however the performance for aliphatic substrates was poor.More recently, Zheng and co-workers demonstrated the use of cobalt as the sole metallic component in TEMPO-catalysed, aerobic alcohol oxidation with a Co(NO3)2/dimethylglyoxime/TEMPO system.65 It was found that weakly-coordinating solvents enabled the best performance with dichloromethane chosen as the optimum solvent. A temperature of 70 °C and 4 bar of O2 was used in these studies. Activated substrates were converted with 1 mol% catalyst loadings and unactivated aliphatic alcohols (primary and secondary) could be oxidised with higher loadings (3 mol% Co and 5 mol% TEMPO).
Vanadium
There are fewer publications that employ vanadium co-catalysts, although the approach developed in 2001 by Neumann and co-workers did make a real impact.66 They reported the combination of TEMPO and the polyoxometalate, H5PV2Mo10O40 for the aerobic oxidation of alcohols. In this case, the polyoxometalate oxidises TEMPO to form the oxoammonium salt which then oxidises alcohols to their corresponding carbonyl compounds. It has also been reported that this catalyst system has been utilised industrially by DSM.5 Despite the fact that this system was used industrially, it has received little research interest (cf. copper and iron systems). In 2012, Lu and co-workers immobilised this polyoxometalate and an ionic liquid functionalised TEMPO on to silica-coated magnetic nanoparticles.67 They demonstrated the oxidation of a range of alcohols and also showed that the catalyst could be separated (using an external magnet) and recycled 10 times without significant loss of activity.Du et al. developed the first example of vanadium as the sole metallic component in TEMPO-catalysed alcohol oxidation with vanadyl sulfate (VOSO4) (4 mol%) and TEMPO (4 mol%) in acetonitrile, O2 (4 bar) at 80 °C.68 The optimised catalyst system oxidised a range of primary benzyl alcohols, with electron-withdrawing substituents on the ring hindering the oxidation process. The secondary alcohol 1-phenylethanol was also easily converted to acetophenone. The catalyst did not perform well with secondary aliphatic alcohols which were only moderately converted; even with extended reaction times (primary aliphatic alcohols were not mentioned).
Laccase
This article mainly focuses on chemo-catalysis; however TEMPO (and derivatives) can be used in conjunction with laccase enzymes. Laccases (EC 1.10.3.2) are multi-copper-containing oxidases that can be used directly for the oxidation of anilines and phenols, or for a wider range of substrates when combined with electron transfer mediators. In 2001 it was shown that TEMPO could act as a mediator for the aerobic oxidation of alcohols.69 It is believed that the mechanism of the laccase/TEMPO system involves the enzyme oxidising the nitroxyl radical to the corresponding oxoammonium cation, which then oxidises the alcohol.70Laccases have several industrial and biotechnological applications;71 however their potential in synthetic chemistry has been less explored compared to the aforementioned chemo-catalytic methods. More detailed discussion on prior work using laccases and mediators for synthetic chemistry can be found in earlier review articles,8,72 while we wish to highlight a very recent addition to the literature. Ying and co-workers reported the benefits of using unhindered nitroxyl radicals as mediators in combination with laccase (from Trametes versicolor).73 In initial screening studies, they tested a range of conditions and nitroxyl radicals; however they primarily studied the use of AZADO. The main advantage of the laccase–AZADO system compared to TEMPO is that it vastly improves the reactivity towards sterically encumbered substrates, as exemplified in Fig. 11.
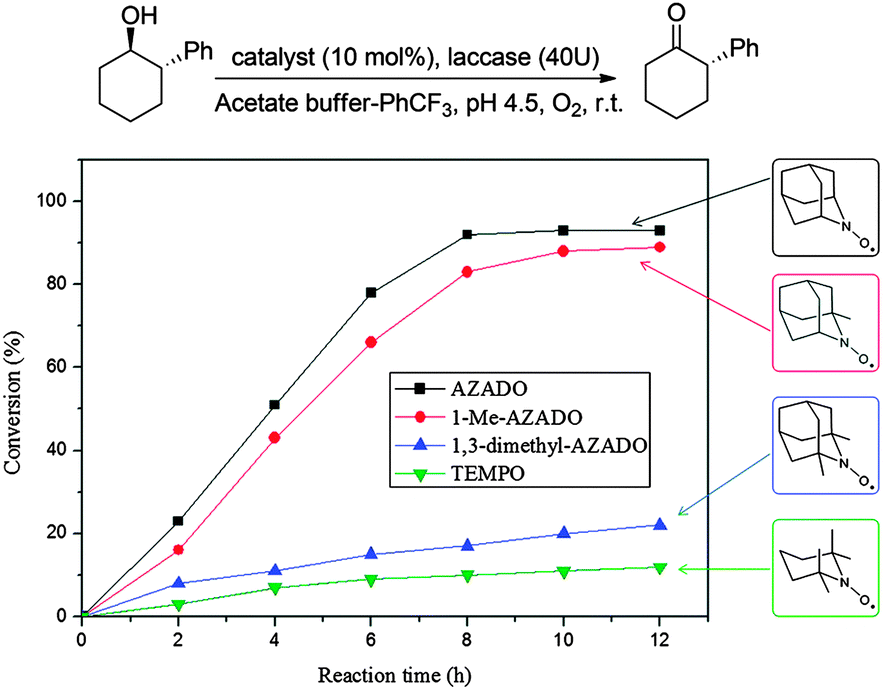 | ||
| Fig. 11 Steric influence of nitroxyl radicals for laccase-nitroxyl catalysed oxidation of alcohols. Reproduced from ref. 73. | ||
For unhindered substrates, there was little difference between AZADO and TEMPO, however for bulkier substrates a significant difference in performance was observed. For example, in the case of menthol, a yield of just 5% was obtained in 36 hours using TEMPO, while AZADO delivered a yield of 84% in 12 hours. They demonstrated that laccase–AZADO could be used for very bulky substrates, including 1-(4-(benzyloxy)-2,6-dimethoxy-3,5-dimethylphenyl)-2-methylbutan-1-ol, which is a precursor for the synthesis of natural products wasabidienone B1 and wasabidienone B0. The system was able to tolerate a range of functional groups, although it did have a few limitations. Laccase oxidised the –OH and –NH2 functionalities in phenol and aniline derivatives leading to the formation of unwanted side products. The system can tolerate thioethers, although substrates with free thiol groups (R–SH) were problematic, as these inhibit laccases.
NOx oxidation systems
The ability to carry out catalytic reactions without the use of any transition metals has a number of potential advantages. For example, as discussed earlier for copper systems, phenols or substrates with chelating ability can bind to the metal and hinder the reaction. There are a number of reports of transition-metal-free aerobic systems with nitroxyl radicals. In these systems, it appears that the oxoammonium salt carries out substrate oxidation and NO2 regenerates the oxoammonium salt. NOx can be generated in situ from nitric acid, nitrates, nitrites or hydroxylamine. As we have already discussed NO2 can readily oxidise nitroxyl radicals to their oxoammonium salts (Fig. 9); however in many of the earlier reports, halides were present suggesting that in such cases bromine, hypobromous acid, nitrosyl bromide or nitrosyl chloride could be acting as active co-oxidants.74 There are clearly similarities between how these systems might operate and the bleach based (i.e. Anelli–Montanari protocol) systems. As mentioned earlier, an issue with such oxidations is that for some substrates there can be selectivity issues (e.g. halogenated by-products). Thankfully, it is possible to operate aerobic NOx systems under halide-free conditions, which avoids such problems.75Early work with NOx systems focused on TEMPO derivatives and although Kakimoto had shown that unhindered radicals such as AZADO resulted in faster reactions, their study still focused on TEMPO.75b Iwabuchi and co-workers demonstrated that unhindered radicals could not only deliver improved reactivity but also a wider substrate scope for such NOx systems.76 They studied a number of AZADO derivatives and found that the best reactivity was obtained with 5-fluoro-2-azaadamantane N-oxyl (5-F-AZADO) (Fig. 12).
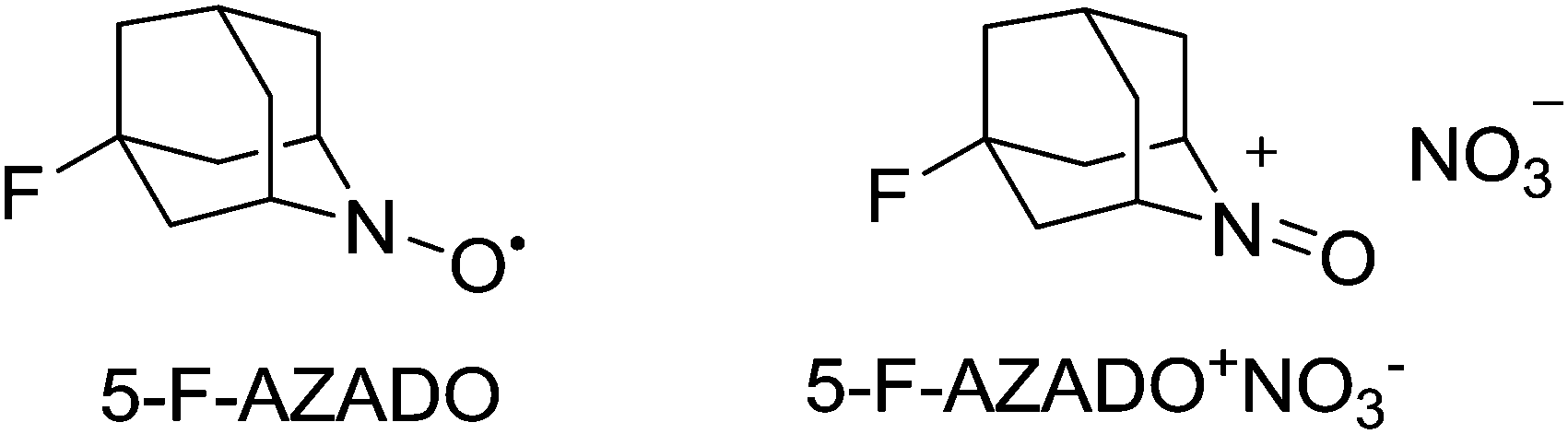 | ||
| Fig. 12 5-F-AZADO and the corresponding oxoammonium nitrate salt. | ||
They postulated that that the increased oxidation potential of 5-F-AZADO compared to AZADO is responsible for the superior reactivity. They also demonstrated that it was possible to oxidise a range of substrates, including bulky substrates, which TEMPO struggles to oxidise. Fig. 13 shows examples of some products that were prepared in this study.
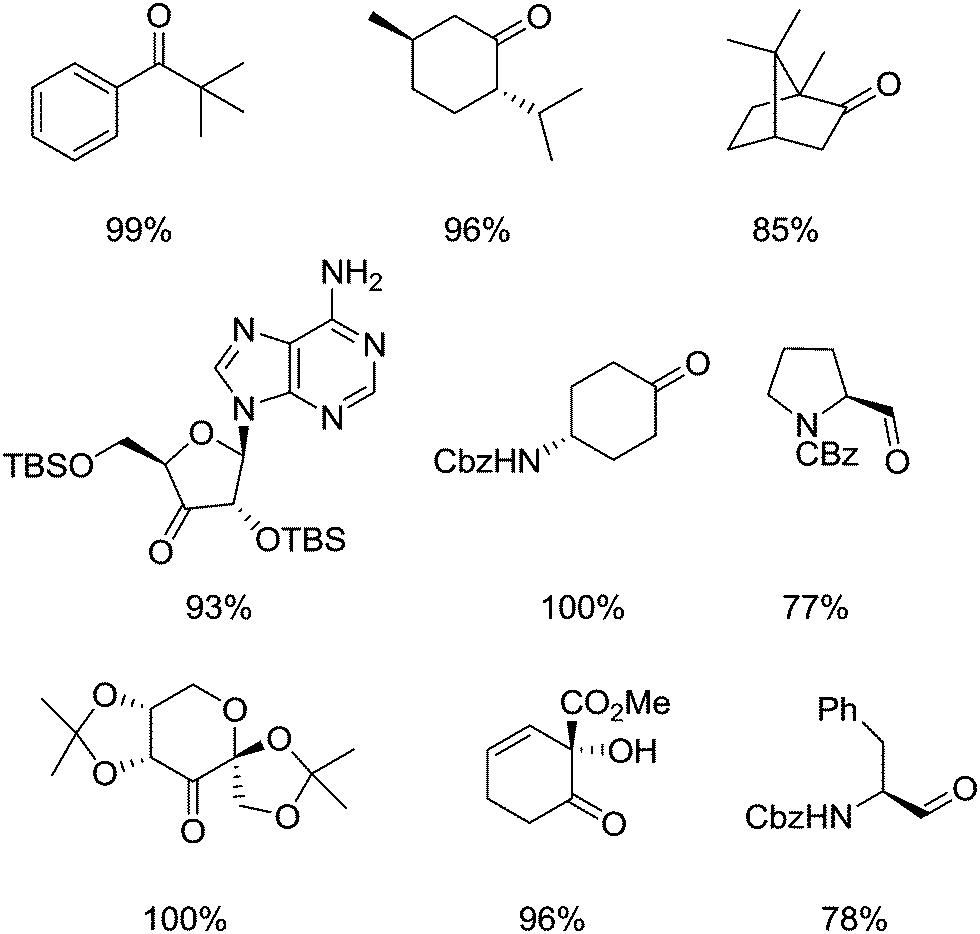 | ||
| Fig. 13 Examples of some products produced via alcohol oxidation using 5-F-AZADO/NaNO2/air or 5-F-AZADO+NO3−/air. | ||
Oxidations were carried out at room temperature and gave high yields in reasonable reaction times (typically a few hours). They also utilised air (balloon) as the terminal oxidant. In general it was shown that 5-F-AZADO (1 mol%) could be used with NaNO2 (10 mol%) or the oxoammonium nitrate salt (5-F-AZADO+NO3−) (Fig. 12) could be formed and used without NaNO2, albeit with a higher catalyst loading (5 mol%). Acetic acid was generally used as the solvent, however, it was demonstrated that by using 5-F-AZADO+NO3− the reactions could be run in acetonitrile with either 2 equivalents of acetic acid or completely acid free; the downside being extended reaction times. A reaction with 5-F-AZADO in dichloromethane was achieved when tert-butyl nitrite (t-BuONO) was employed. They also showed that it was also possible to precipitate 5-F-AZADO+NO3− at the end of the reaction (72% was recovered) with the recovered salt showing the same reactivity in a subsequent reaction. Indeed, around a similar time, Kakimoto and co-workers reported how 1-methyl-2-azaadamantane N-oxyl (1-Me-AZADO) could be recycled.77 This radical was used in conjunction with NaNO2 and HNO3 and the salt 1-Me-AZADO+NO3− was separated from the organic (product) phase due to its water solubility and then re-used.
More recently Lauber and Stahl studied the use of ABNO and ketoABNO for such NOx type systems.78 Although the previous work using AZADO based radicals demonstrated good performance, this more recent study was motivated by the fact that ABNO based radicals are significantly easier to prepare. In this study they initially prepared a range of ABNO derivatives, many of which had not been reported previously. These ABNO based radicals were then compared along with a number of TEMPO derivatives for their ability to oxidise cyclohexanol. It was found that ABNO and ketoABNO delivered the best performance in these screening tests. Interestingly, the results did not show a correlation between redox potential and catalytic performance. The authors said efforts to understand the factors behind good catalyst performance were underway, but suggested that in some cases the derivatives may not be as stable under the reaction conditions. In this study, they then examined ABNO and ketoABNO for a range of substrates, using three different methods. (Fig. 14)
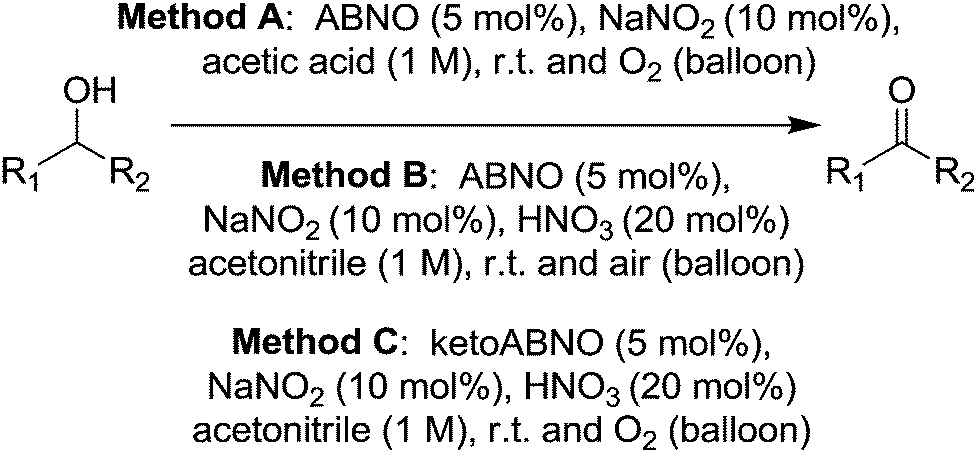 | ||
| Fig. 14 Methods for alcohol oxidation using ABNO or ketoABNO. | ||
Excellent yields could be obtained for a range of functionally diverse substrates. Method A gave the best performance overall, being applicable to the widest range of substrates, although in some cases Method C delivered higher yields. Although they demonstrated that these nitroxyl NOx systems have excellent substrate scope, they also highlighted that there are some limitations. For example aniline derivatives are not suitable as the conditions may lead to the formation of diazo compounds. They also found that tertiary amines could be low yielding and a possible explanation for this was that such substrates could react with the acids that are utilised in these methods.
There are a number of papers which have looked at immobilising radicals for NOx systems to facilitate the recycling of the radical. Zhu et al. utilised a TEMPO functionalised ionic liquid with NaNO2 and the radical could be separated and recycled.79 As part of their studies examining a system which used NH4NO3 as the source of NOx, Stavber and co-workers employed a commercially available polymer-supported form of TEMPO, enabling the radical to be recycled.80 Karimi et al. have developed recyclable systems that use TEMPO covalently tethered to mesoporous silica (SBA-15) in conjunction with either sodium nitrite81 or t-BuONO.82 They also reported a t-BuONO system with TEMPO supported on magnetic core–shell nanoparticles, which can be easily separated (magnetically) and re-used.83 There have also been studies,84 including mechanistic studies,85 where NOx gas has been adsorbed onto silica-tethered TEMPO and then used for alcohol oxidation.
The most efficient way to utilise such solid supported catalysts is to operate a continuous flow system, something that was recently reported by Hermans and co-workers.86 They demonstrated the aerobic oxidation of alcohols using (commercially available) silica-immobilised TEMPO and with catalytic amounts of HNO3 as the source of NOx. As mentioned earlier, flow systems also offer improved safety and in this case 5 bar of O2 was used. They demonstrated that this flow approach had superior space-time-yields (i.e. the product yield per unit of time and per reactor volume) compared to previously reported batch systems. The flow system was tested on a number of representative model substrates including aliphatic alcohols and renewable substrates (lactic acid and 5-hydroxymethylfurfural).
In summary, the recent developments in stable radicals for aerobic oxidation of alcohols are very promising. It has been shown that these systems have excellent substrate scope and tolerance of a wide range of functional groups. Each method has specific substrate limitations, but the correct choice of method should allow most substrates to be oxidised efficiently. The methods often employ very mild conditions and it has been shown in some cases that it is possible to combine these oxidation reactions with other reactions in a one-pot manner. For example, Christmann and co-workers have described the Cu(I)/TEMPO oxidation of Z-allylic alcohols or E/Z mixtures with concomitant isomerisation (by DMAP) to form (E)-α,β-unsaturated aldehydes.87 They also demonstrated that alcohol oxidation followed by an organocatalytic intramolecular Diels–Alder reaction could be carried out in one-pot, while Jang and co-workers demonstrated that a Cu/TEMPO oxidation of allylic alcohols could be carried out in tandem with the enantioselective Michael addition using a chiral amine catalyst.88 They also found that if TEMPO was added in larger quantities, an addition reaction took place, resulting in tandem oxidation/Michael addition/α-oxyamination reactions. There are a number of examples utilising these catalyst systems for the Passerini reaction (Fig. 15).89 This reaction involves the condensation of an isocyanide with an aldehyde or ketone and carboxylic acid. These studies demonstrated that aldehydes or ketones could be generated in situ from the oxidation of alcohols.
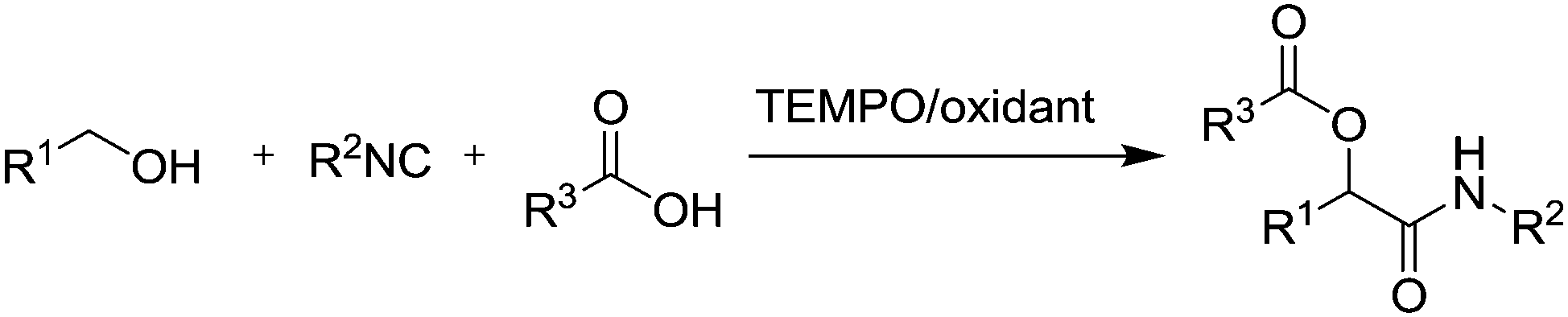 | ||
| Fig. 15 Passerini reaction using alcohols. | ||
In the studies we have highlighted, model substrates have been examined that are of relevance to applications in the fine chemical, pharmaceutical and agrichemical industries. A number of other studies have explored the oxidation of lignin and lignin model compounds, an important challenge if we are to exploit renewable resources for chemicals. These reactions involve alcohol oxidation and also C–C bond cleavage reactions. Hanson and co-workers have examined Cu/TEMPO catalyst systems,90 while Stahl and co-workers examined a range of catalysts systems, focusing on the use 4-acetamido-TEMPO (4-AcNH-TEMPO) combined with HNO3, and HCl as co-catalysts.91 The results from these studies are promising, and stable radicals may have a role to play in this extremely difficult challenge.
Expanding the scope of reactions
Although the majority of studies reported to date have examined alcohol oxidation, in recent years there has been a growing number of publications looking at other oxidative transformations. Herein we will highlight some of the recent developments involving stable radicals.C–N bond formation
There have been a number of interesting reports of C–N bond forming reactions using TEMPO based catalysts. Zhang and Jiao reported a novel Cu-catalysed oxidative amidation–diketonisation reaction of terminal alkynes leading to α-ketoamides, in which molecular oxygen is used as both the oxidant and a reactant (Fig. 16).92 Labelling studies indicated the product oxygen atoms came from O2 rather than H2O. The reaction was mainly limited to activated substrates, for example an aliphatic alkyne was low yielding and aliphatic amines were unreactive.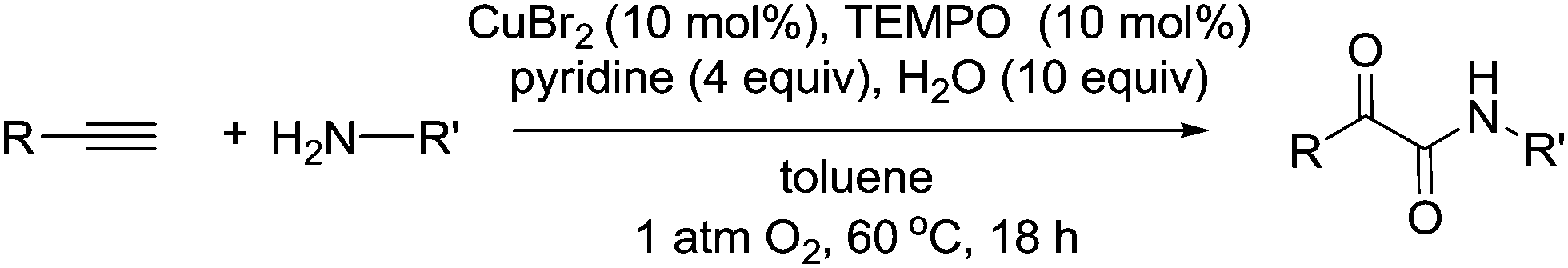 | ||
| Fig. 16 The formation of α-ketoamides from aromatic amines and terminal alkynes. | ||
More recently Chen and co-workers reported the use of iron nitrate/TEMPO for the preparation of amides from aromatic alcohols and amine hydrochloride salts.93 This was a one pot, two-step procedure whereby Fe(NO3)3/TEMPO was used to carry out aerobic oxidation of the alcohol (limited to simple aromatic alcohols). Once the aldehyde was produced, the (aliphatic) amine hydrochloride salt was added along with CaCO3 and tert-butyl hydroperoxide (TBHP). In this second step, it is believed that the oxidation of the hemiaminal is performed by Fe(III) and TBHP.
It is also worth mentioning a method that was reported in 2010 for the intermolecular C–H bond amination of acidic aryl C–H bonds with primary aromatic amines.94 This was applied to electron-deficient polyfluoroarenes and polychloroarenes and azoles as shown in Fig. 17. The role of TEMPO was unclear, however its presence (at relatively high loadings) did improve the product yields.
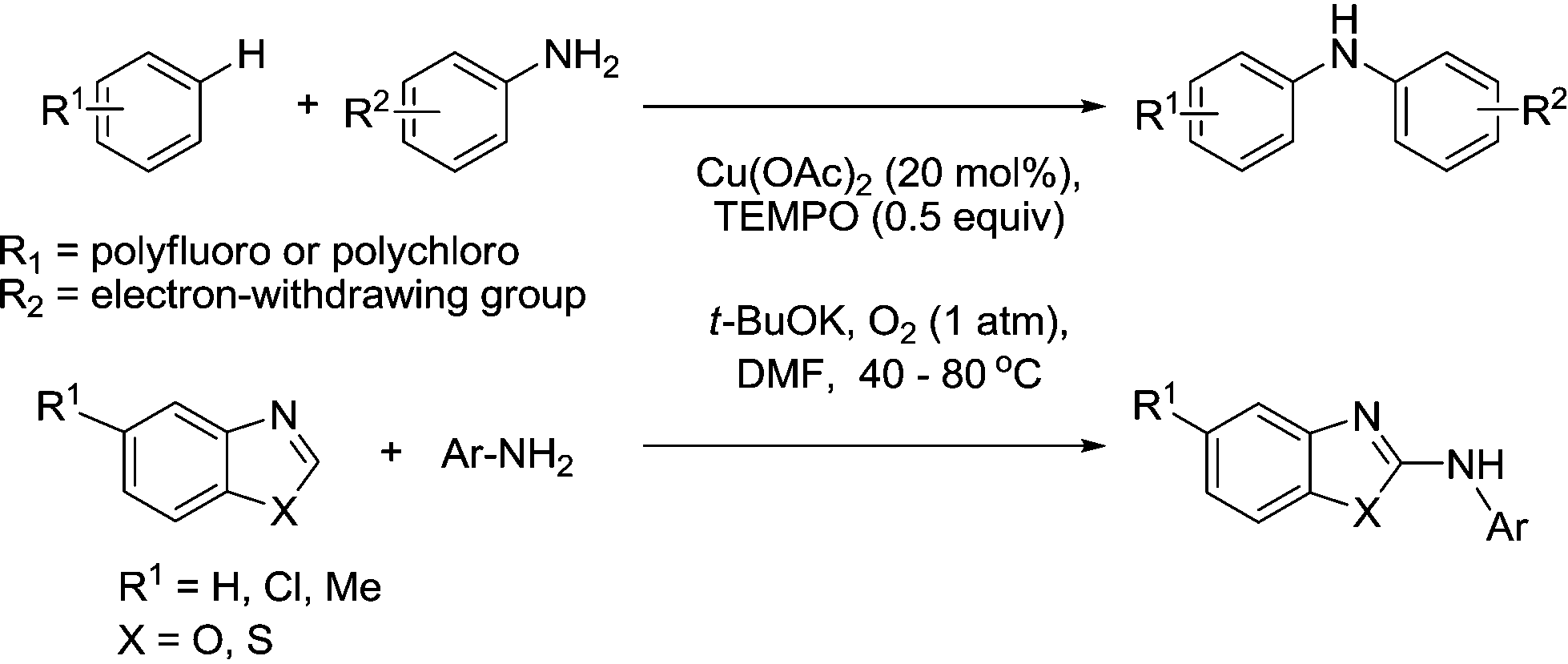 | ||
| Fig. 17 Cu/TEMPO C–H bond amination. | ||
Maiti and co-workers reported methods for the stereoselective nitration of olefins. In their first report, they combined TEMPO (0.4 equiv.) with AgNO2 (3 equiv.) to carry out the nitration of a wide range of olefins with selectivity to the E isomer.95 The method was then improved when they demonstrated that it was possible to avoid the use of silver salts (Fig. 18). First they showed that Fe(NO3)3 could be used,96 then they reported a metal-free protocol using t-BuONO.97
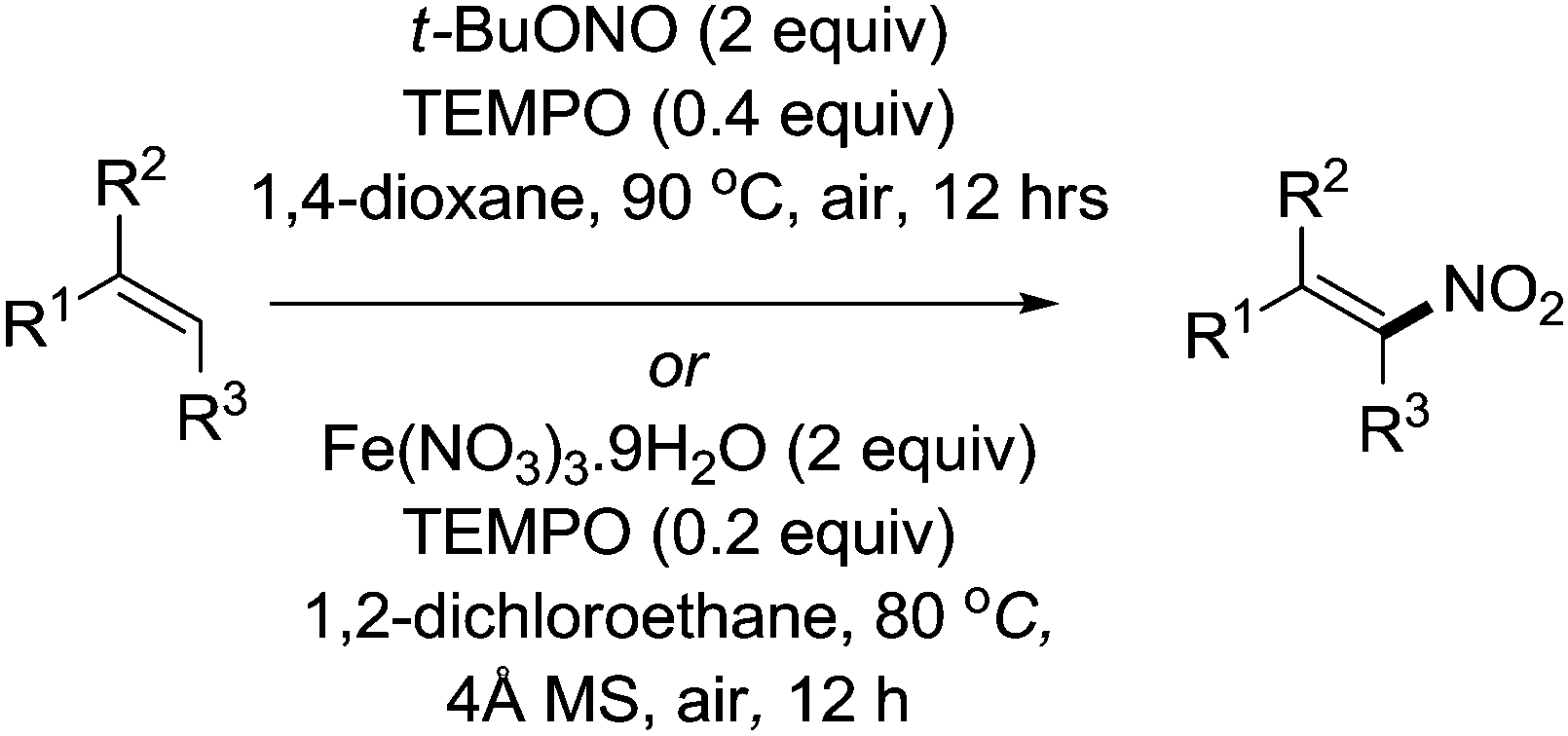 | ||
| Fig. 18 Stereoselective nitration of olefins. | ||
In these studies they demonstrated the utility of these methods for a wide variety of substrates: aromatic, aliphatic, hetero-aromatic and even substrates containing other alkene or alkyne functionality. In most cases, the products were obtained in good to excellent yields and had excellent E selectivity. Alkenes close to electron withdrawing groups reacted slower; therefore aliphatic terminal olefins could be selectively nitrated over a terminal alkene or indeed alkyne next to an ester group. There were also examples of substrates where a terminal olefin could be selectively nitrated in the presence of an internal olefin.
Maiti and co-workers also reported the (E)-nitroolefins via the decarboxylative nitration of α,β-unsaturated carboxylic acids, using t-BuONO (2 equiv.) and TEMPO (80 mol%).98 It was shown that such reactions could proceed without TEMPO but a mixture of E and Z isomers were obtained, while TEMPO led to selective formation of the E isomer.
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) N bond formation
N bond formation
Synthesis of imines
Imines are versatile intermediates and can be used to prepare a variety of important chemicals. They are typically prepared from the condensation of amines with carbonyls (usually aldehydes). Oxidation catalysts can be used to make imines from alcohols and amines, in other words, generating the carbonyl in situ using an oxidation reaction. Imines can also be prepared via dehydrogenation of amines.99Fig. 19 outlines the ways in which metal/nitroxyl radical catalysts have been used for the synthesis of imines (and azo compounds).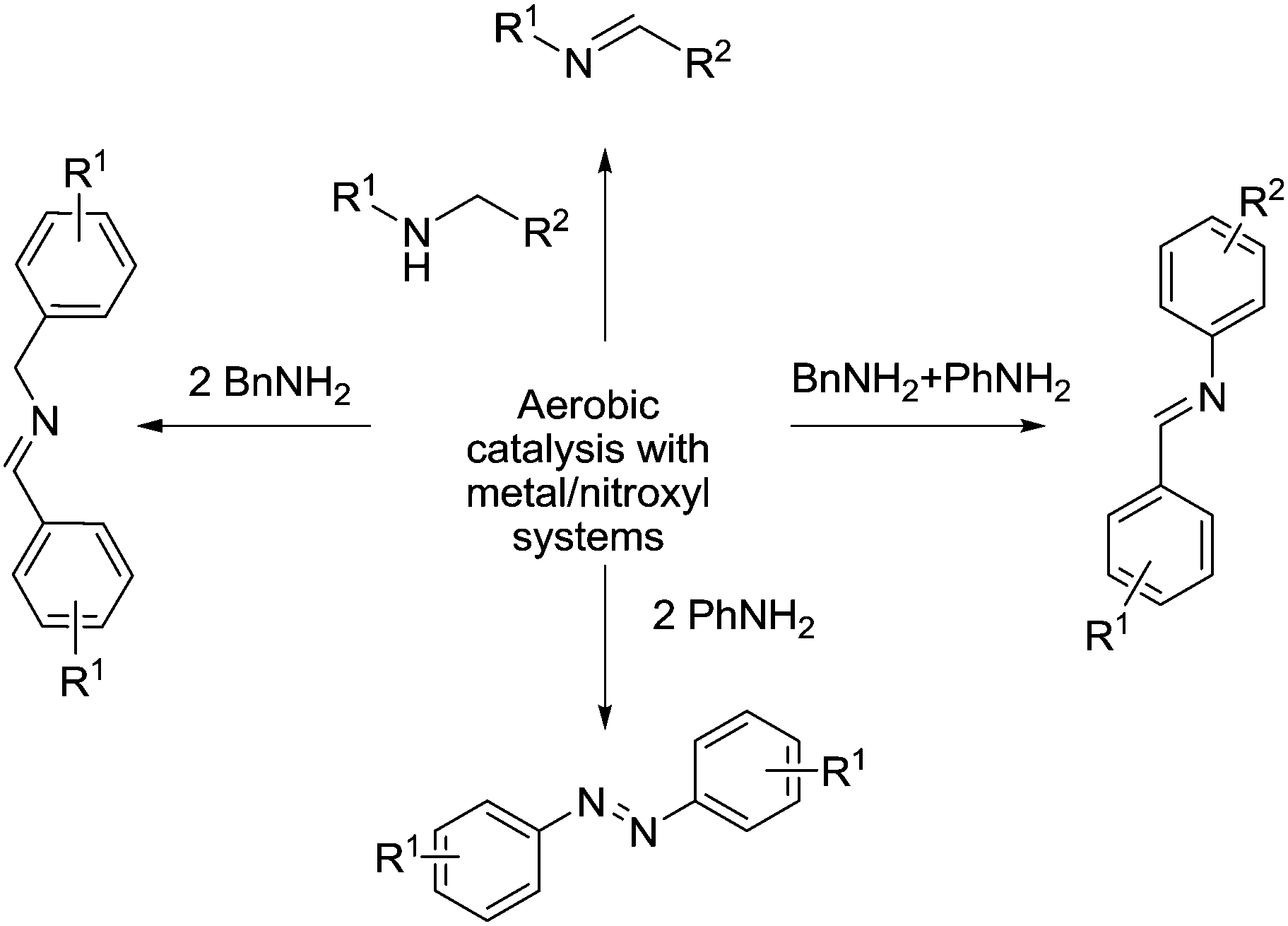 | ||
| Fig. 19 General overview of the synthesis of imines and azo compounds via metal/nitroxyl/O2 catalysis. | ||
In 2011, Xu and co-workers described a mild one-pot imine synthesis from alcohols and amines with Pd(OAc) and TEMPO.100 The reaction proceeded without TEMPO however superior performance was obtained when the radical was present, although the reason for this is not clear.
In 2012, the same group reported that Cu/TEMPO could be used to prepare imines via this route. As discussed earlier, the replacement of Pd with Cu is desirable and it was found that Cu/TEMPO could be used to prepare a wide variety of structurally diverse imines in good to excellent yields.101 It also operated under very mild conditions; CuI (1 mol%), bpy (1 mol%) and TEMPO (2 mol%) in CH3CN at room temperature and with an “open flask”. Although aldehydes react with amines to form imines in the absence of a catalyst, it was also shown in this paper that CuI accelerates this condensation reaction; consequently this one-pot approach from alcohols is a sensible route for the preparation of imines.
In 2011, Hu and Kerton demonstrated that CuBr2/TEMPO could be used to prepare imines from benzylamines under air at 25 °C in CH3CN–H2O.102 The catalyst could also be used for the oxidative coupling reaction between benzylamines and anilines at 45 °C. They also showed that electron donating anilines could undergo dehydrogenative coupling to form azo compounds in good yields at 60 °C. As discussed earlier, Cu(I) systems are more active for alcohol oxidation, and recently Xu and co-workers reported a copper(I)/TEMPO system for the oxidation of amines to imines under neat conditions (i.e. no additional solvent).103 The same group also reported the use of Fe(NO3)3/TEMPO for the synthesis of imines.104 In this study they demonstrated that this catalyst could be used for the synthesis of imines via the oxidation of amines, the oxidation of alcohols with amines and also the oxidation of anilines with benzylamines to form cross imines. They showed a broad substrate scope, however aliphatic substrates were not suitable for this catalyst system.
In 2012, Sonobe et al. published an excellent paper which described the synthesis of imines and their subsequent reactivity in C–C bond forming reactions.105 This study utilised copper(I) complexes in combination with the sterically unhindered nitroxyl ketoABNO. Significantly this catalyst system was able to oxidise aliphatic amines and bulky amines that were previously not suitable when TEMPO was employed. It was clear that the utilisation of ketoABNO made possible reactions that could not be carried out using TEMPO. For example, they employed bulky ligands to ligate the copper and this would not have been suitable with the sterically hindered TEMPO. We will highlight the C–C bond forming reactions shown in this study in a later section.
Oximes and oxime ethers
In 2012, Wertz and Studer reported a metal free TEMPO catalysed method for preparing oximes and oxime ethers from hydroxylamines and alkoxyamines (Fig. 20).106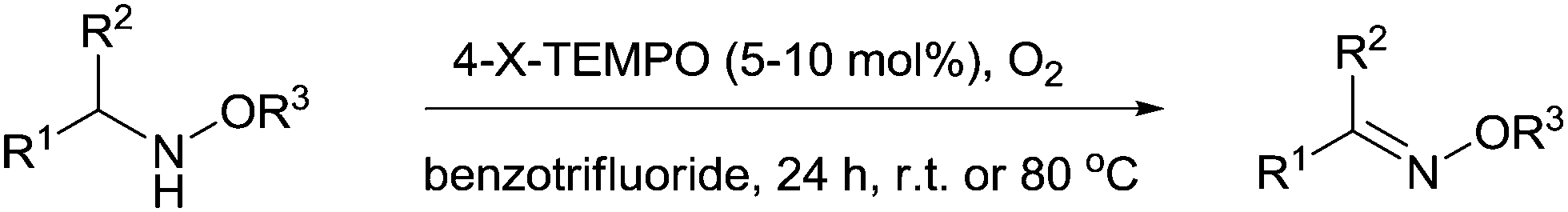 | ||
| Fig. 20 TEMPO mediated oxidation of hydroxylamines and alkoxyamines to oximes and oxime ethers. | ||
They utilised TEMPO, 4-OH-TEMPO or 4-acetamido-TEMPO (4-AcNH-TEMPO) for these reactions and while these all performed similarly, the 4-substituted radicals were more readily separated from the product compared to TEMPO. It was found that TEMPO (or the derivatives) could not act catalytically for aliphatic hydroxylamines, which were less reactive and in those cases an excess of TEMPO was required. For alkoxyamines and benzylic hydroxylamines it was possible to use catalytic amounts of the radical along with O2 to produce products in good to excellent yields. They also showed that benzyl bromides could be converted to oxime ethers in a one-pot procedure where the alkoxyamines were formed in situ, although excess TEMPO (2.2 equivalents) was required in this case.
Suzuki et al. outlined a method for the aerobic oxidation of amines to oximes.107 This is a rare example that does not utilise a nitroxyl radical as the stable radical. Here they have used 1,1-diphenyl-2-picrylhydrazyl (DPPH) in combination with a heterogeneous tungsten (WO3/Al2O3) co-catalyst (Fig. 21). The catalyst could oxidise alicylic and aliphatic amines to their corresponding oximes in excellent yields. The catalyst showed good selectivity for amines. For substrates that possessed alcohol groups (e.g. 5-hydroxylpentylamine and 4-hydroxycyclohexylamine) the amine could be oxidised and the alcohol group remained intact. They also demonstrated that the DPPH–WO3/Al2O3 catalyst could be recycled and reused (3 cycles) without loss of performance. In the case of secondary amines, it was possible to oxidise these to their corresponding nitrones. In these studies, they used a pressurised reactor and a 7% O2 gas mixture to ensure reactions were outside the flammability limits.
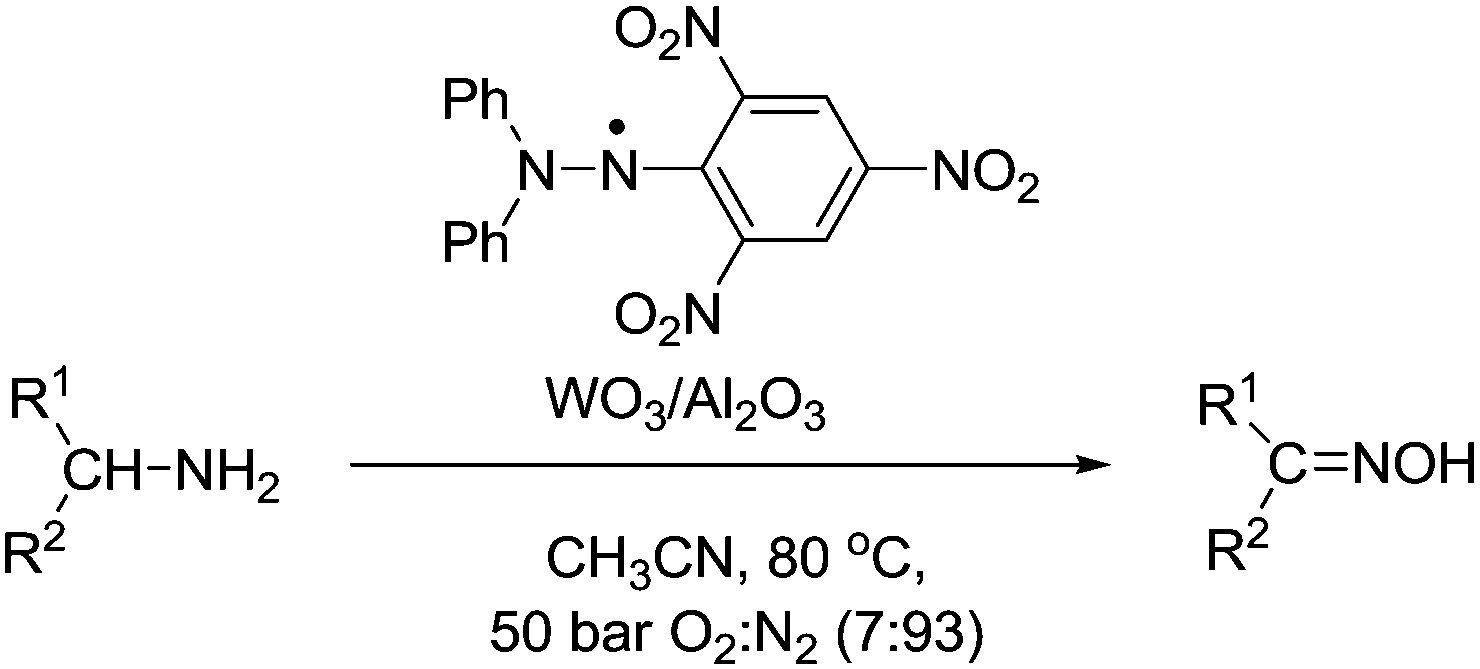 | ||
| Fig. 21 Aerobic oxidation of amines to oximes using DPPH and WO3/Al2O3. | ||
Synthesis of N-heterocycles
Nitrogen containing heterocycles are often the basis of natural products and pharmaceuticals, therefore efficient methods to produce such structures are valuable. Han and co-workers have published a number of papers describing the synthesis of N-heterocycles employing TEMPO and TEMPO derivatives as catalysts. In 2008, they reported that 4-methoxy-TEMPO could be utilised as a catalyst for the synthesis of 2-substituted benzoxazoles, benzothiazoles, and benzimidazoles utilising the route shown in Fig. 22.108 They prepared a range of substituted heterocycles in fair to excellent yields.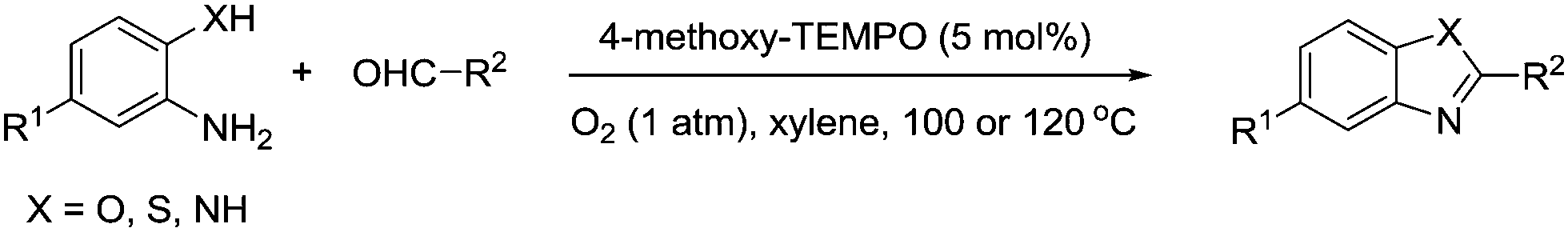 | ||
| Fig. 22 Synthesis of benzoxazoles, benzothiazoles, and benzimidazoles using 4-methoxy-TEMPO. | ||
In 2011, they described the synthesis of 2-aryl quinazolines by reacting arylmethanamines with either 2-aminobenzoketones or 2-aminobenzaldehydes (Fig. 23).109 Once again this was a metal-free system but they employed 4-hydroxy-TEMPO as the catalyst in this instance. Products were obtained in good to excellent yields.
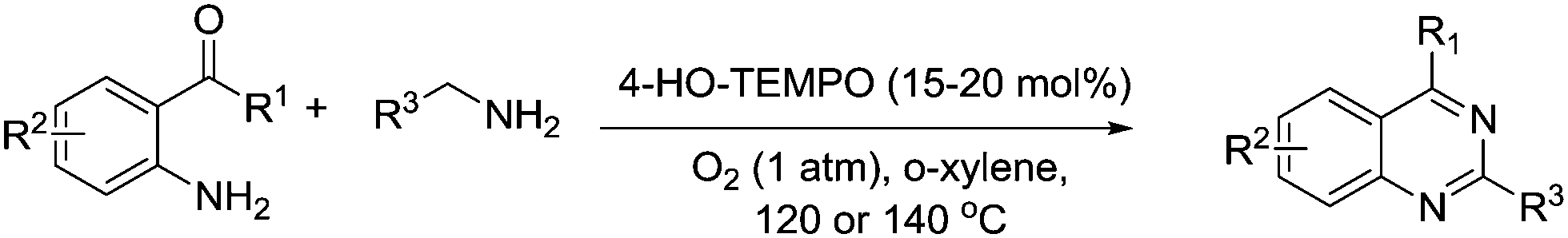 | ||
| Fig. 23 Synthesis of 2-aryl quinazolines using 4-HO-TEMPO. | ||
In 2012, they reported another method for the synthesis of 2-substituted quinazolines and 4H-3,1-benzoxazines and in this case, they utilised a catalyst system more similar to that employed for alcohol oxidation.110 Screening various conditions it was found that the addition of CuCl improved the yield and rate of these reactions. It was also noted that the addition of nitrogen ligands had a positive effect with 4-diazabicyclo[2.2.2]octane (DABCO) delivering the best performance, with optimised conditions shown in Fig. 24. The optimised conditions used lower loadings of 4-hydroxy-TEMPO and lower temperatures compared to their previous study. A range of substituted heterocycles were prepared in good to excellent yields for the majority of cases.
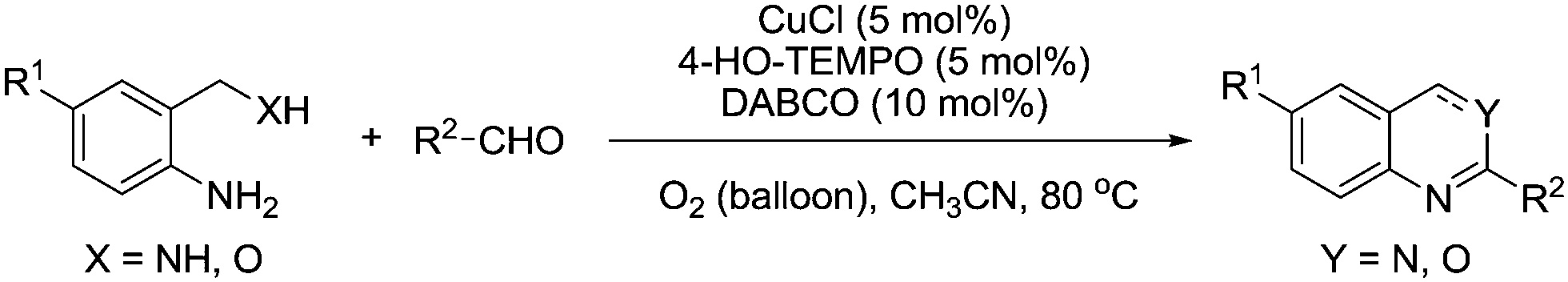 | ||
| Fig. 24 Synthesis of 2-substituted quinazolines and 4H-3,1-benzoxazines using Cu/4-HO-TEMPO. | ||
In 2013, Chen et al. developed a three-component cascade synthesis of quinazoline derivatives using CuCl, bpy and TEMPO in the presence of cerium nitrate (Fig. 25).111 (2-Aminophenyl)methanols and a variety of substituted benzaldehydes were converted smoothly to their corresponding 2-arylquinazolines in moderate to excellent yields.
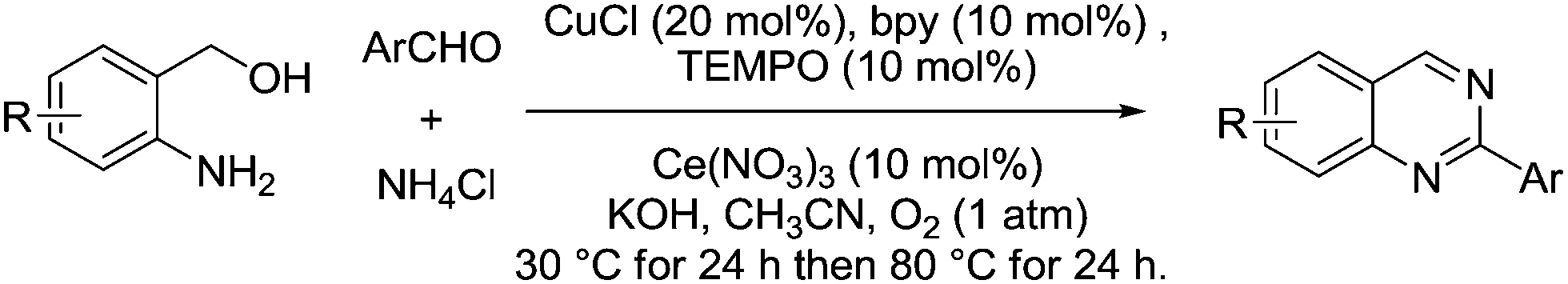 | ||
| Fig. 25 Synthesis of quinazoline derivatives from (2-aminophenyl)methanols, aldehydes and ammonium chloride. | ||
In 2012, we reported a method for synthesising indoles and quinolines via aerobic oxidation of amino alcohols.112 As shown in Fig. 26, we utilised the Cu/TEMPO catalyst system previously optimised by Kumpulainen and Koskinen.29
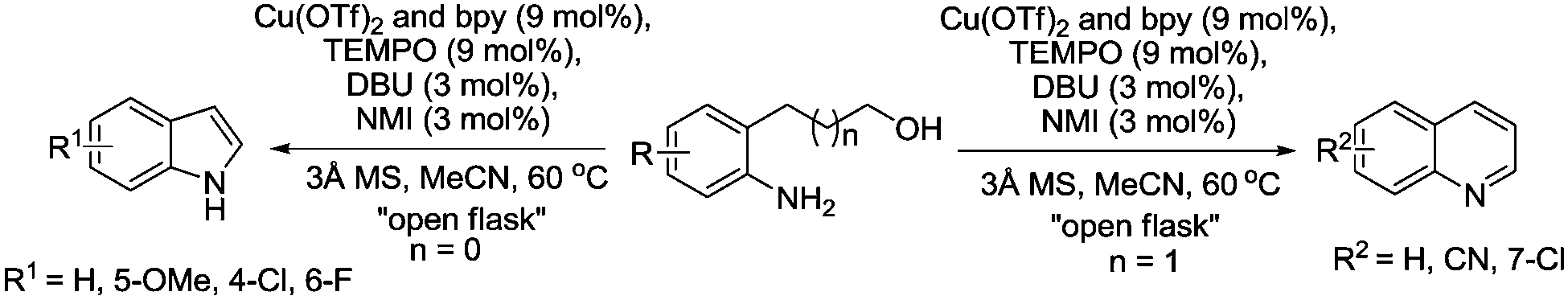 | ||
| Fig. 26 Synthesis of N-heterocycles via Cu/TEMPO catalysed aerobic oxidation of amino alcohols. | ||
This study differs from the previous examples by Han and co-workers because in this case, alcohol oxidation is a key step in the reaction. Once the aldehyde is formed an intra-molecular cyclisation and dehydration takes place to form the heterocycle. Indoles were prepared in moderate to good yields, although it was found that extended reaction times led to poorer yields and control experiments indicated that the catalyst caused product decomposition. The selectivity of Cu/TEMPO for primary alcohols was demonstrated using a substrate containing a secondary alcohol group, which remained intact. For the synthesis of quinolones it was found that 1,2,3,4-tetrahydroquinoline-2-one was formed as by-product, indicating that upon cyclisation there were two competing pathways; dehydration or further alcohol oxidation to give 1,2,3,4-tetrahydroquinoline-2-one. The use of molecular sieves and a drying tube helped promote the synthesis of the desired quinoline.
In 2013, Li and co-workers developed a room temperature oxidative aromatisation of substituted Hantzsch 1,4-dihydropyridines (1,4-DHPs) using either FeCl3 or CuCl2 combined with NaNO2 and TEMPO (Fig. 27).113 In most cases, FeCl3 was used although there were some examples where CuCl2 gave better performance. It was possible to obtain a range of products in excellent yields in under 60 min.
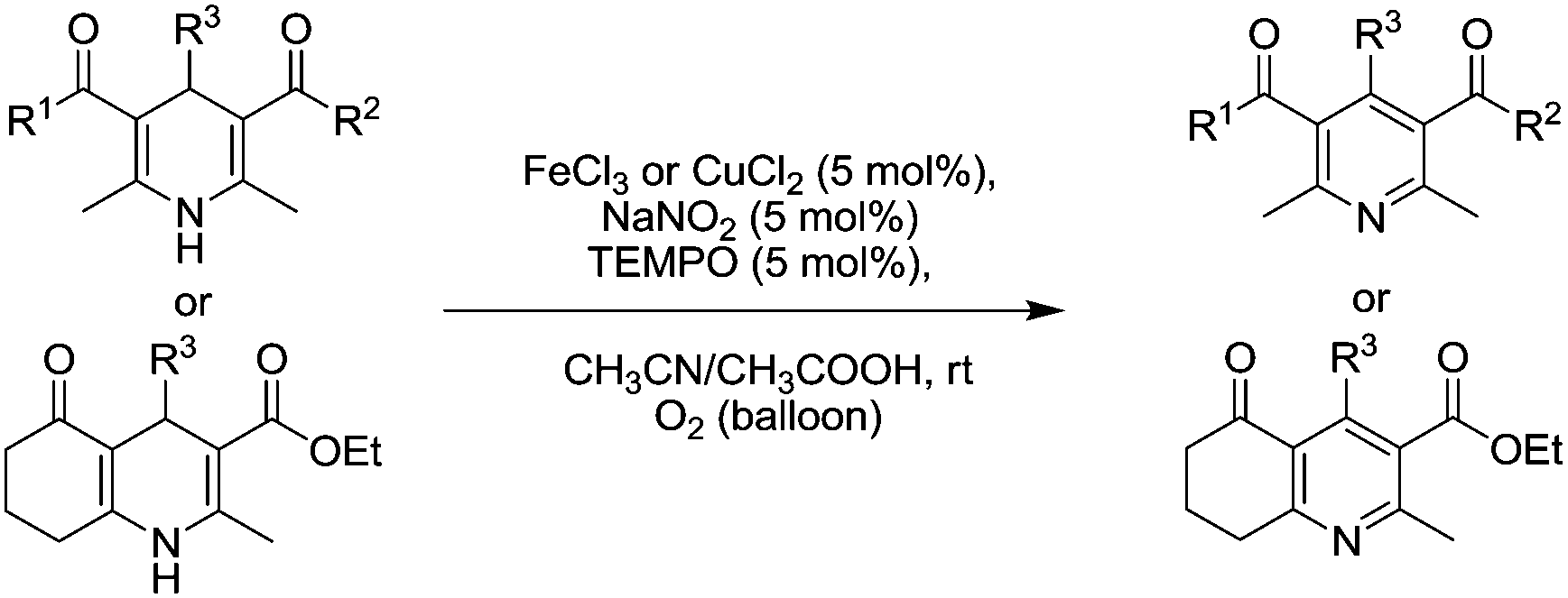 | ||
| Fig. 27 oxidative aromatisation of substituted Hantzsch 1,4-dihydropyridines (1,4-DHPs). | ||
Whilst this review focuses on the use of stable radicals as catalysts, it is worth mentioning related work in the synthesis of heterocycles. Mancheño and co-workers have utilised TEMPO derived oxoammonium salts as a stoichiometric oxidant for the synthesis of oxazinones,114 and substituted quinolines and dihydroquinazolines.115,116 While Chiba and co-workers reported the synthesis of substituted isoxazoles and pyrazoles, via TEMPO mediated C–H bond oxidation of oximes and hydrazones, although excess quantities of TEMPO (3–6 equiv.) were employed in this example.117
C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) N bond formation
N bond formation
Very recently, there were four independent reports (including one from our group), published within a short space of time, demonstrating the use of aerobic Cu/nitroxyl systems for the synthesis of nitriles.118–121 The nitrile group is an important functionality in chemistry and often the synthesis of nitriles relies on undesirable methods. In 1983, Semmelhack and Schmid reported that TEMPO could be employed for the electro-oxidation of amines to nitriles;122 however there were no aerobic methods for selectively producing nitriles using such radicals. In these recent publications, three papers focused on the synthesis of nitriles from alcohols or aldehydes using aqueous ammonia as the nitrogen source (Fig. 28). Starting from alcohols, the catalyst produces the aldehyde and subsequently oxidises the imine, which readily forms via the aldehyde and ammonia.
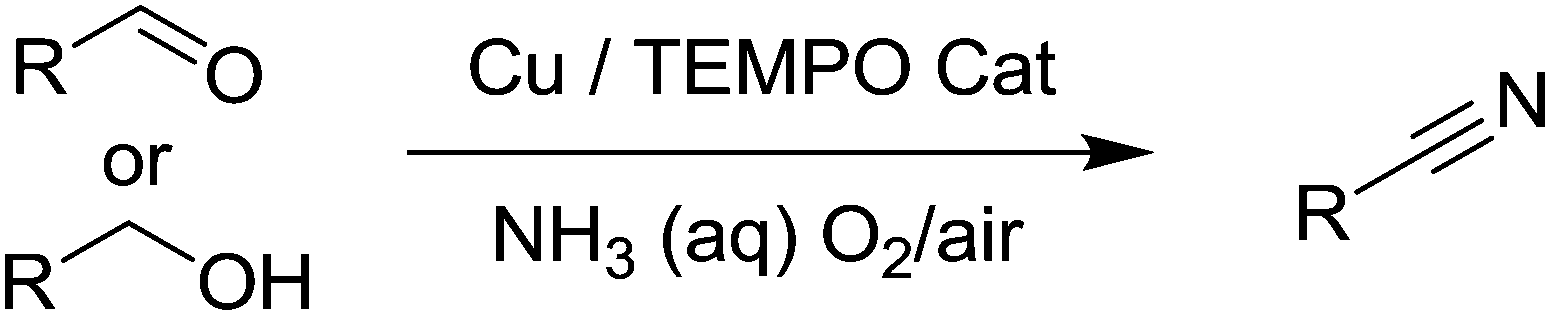 | ||
| Fig. 28 Synthesis of nitriles from alcohols or aldehydes using Cu/TEMPO/O2 systems. | ||
Tao and co-workers used 5 mol% Cu(NO3)2 (no ligand was used), 5 mol% TEMPO, 3 equivalents of NH3 and DMSO as the solvent. The flask was flushed with O2 and then sealed and heated to 80 °C for 5 hours. For aldehydes the reaction could be performed without TEMPO. A range of benzylic substrates were demonstrated but the catalyst was not suitable for aliphatic substrates. They also demonstrated that a one-pot procedure could be employed for the synthesis of aryl amides from the corresponding benzyl alcohol. Once the nitrile had been produced, acetaldoximine and water were then added to the reaction mixture and heated to 100 °C for 24 hours.
Huang and co-workers described a method that utilised a more reactive Cu(I) system. This enabled a wide range of functionalised alcohols (including aliphatic) to be converted to the corresponding nitrile, in good to excellent yields, under mild conditions. They utilised a catalyst system composed of 5 mol% CuI, 5 mol% bpy and 5 mol% TEMPO. They added 2 equivalents of aqueous ammonia and carried out the reaction under an O2 atmosphere (balloon). Activated alcohols were converted at room temperature (24 hours) with ethanol as the solvent. Reactions with aliphatic substrates were carried out in acetonitrile at 50 °C (24 hours). In the case of benzylic alcohols they also demonstrated that a one-pot procedure could be employed for the synthesis of biaryl heterocycles. Once the nitrile was produced reagents were then added to convert the nitrile to tetrazoles, imidazolines, thiazolidines, triazolopyridines and oxazolines.
We avoided the use of pure O2 atmospheres and developed methods that used air (open flask) or dilute oxygen (8%) mixtures. Activated aldehydes were converted to the corresponding nitrile using an “open flask” method. 10 mol% Cu(OTf)2, bpy and TEMPO were used. The water soluble radical 4-sulfonatooxy-TEMPO was also used as this could be readily washed out from the product. The reaction was carried out in acetonitrile–water and NaOH, with the presence of this base improving the reaction rate. This system was open to the air and therefore aqueous ammonia was slowly added during the reaction to replace ammonia that had evaporated. The “open flask” method could be used from 25–70 °C, above this temperature the ammonia evaporated before reacting. It was found that aliphatic substrates were slow to react using this method, therefore based on Stahl's Cu(I) studies,30 we switched to using [Cu(MeCN)4][OTf]. The improved reactivity of Cu(I)/TEMPO is not maintained when substantial amounts of water are present, therefore acetonitrile was used along with 2.5 equivalents ammonia. To prevent evaporation of ammonia, a balloon containing air was attached to the condenser. The modified method allowed octanal to be converted cleanly at 25 °C while 1-octanol required 50 °C. With an aromatic aldehyde at a temperature of 120 °C, inexpensive CuCl2 could be used at 1 mol% along with 1 mol% 4-sulfonatooxy-TEMPO. At such a temperature, a pressurised reactor is required to prevent ammonia evaporation; therefore a pressurised atmosphere of 8% O2 was used.
Kim and Stahl had also found that nitriles could be produced from alcohols and ammonia; however due to the aforementioned studies they focused on the selective oxidation of primary amines to nitriles. Oxidation of amines can lead to a variety of products,123 and as discussed earlier Cu/TEMPO catalyst systems can catalyse the aerobic oxidation of primary amines to the corresponding homocoupled imines. In those studies nitriles are sometimes seen as a by-product.102,103 Kim and Stahl demonstrated that unhindered radicals ABNO and AZADO improved the yield and selectivity of nitrile versus homocoupled imine. As shown in Fig. 29 the optimised conditions utilised 4,4′-tBu2bpy (4,4′-di-tert-butyl-2,2′-bipyridyl) as a ligand, DMAP (4-dimethylaminopyridine) as the base and ABNO as the radical.
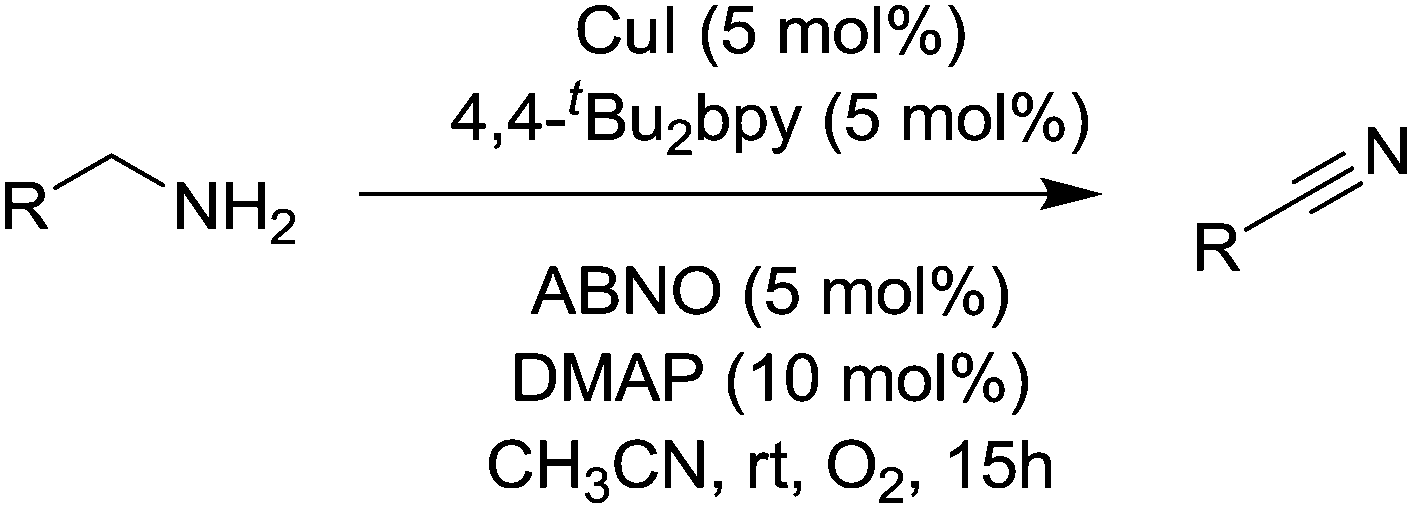 | ||
| Fig. 29 Aerobic oxidation of primary amines to nitriles. | ||
This catalyst system was able to convert a range of benzylic, allylic and aliphatic primary amines to nitriles, in good to excellent yields. Preliminary mechanistic experiments indicate that the nitrile is not formed via the homocoupled imine, but rather that catalyst carries out a double oxidation; oxidation of the amine to the primary imine followed by oxidation of this imine to the nitrile.
Carbon–carbon bond formation
The formation of C–C bonds is a powerful tool in organic synthesis. Recently, there have been several reports that have utilised stable radicals for the construction of C–C bonds, using a number of different approaches.In 2008, Studer and co-workers reported the use of TEMPO as an aerobic catalyst for the homocoupling reaction of aryl-, alkenyl- and alkynyl-Grignard reagents.124 More recently, they followed this up with a study showing that TEMPO could be used as a catalyst for the cross-coupling reaction between nitrones and alkynyl-Grignard reagents (Fig. 30).125
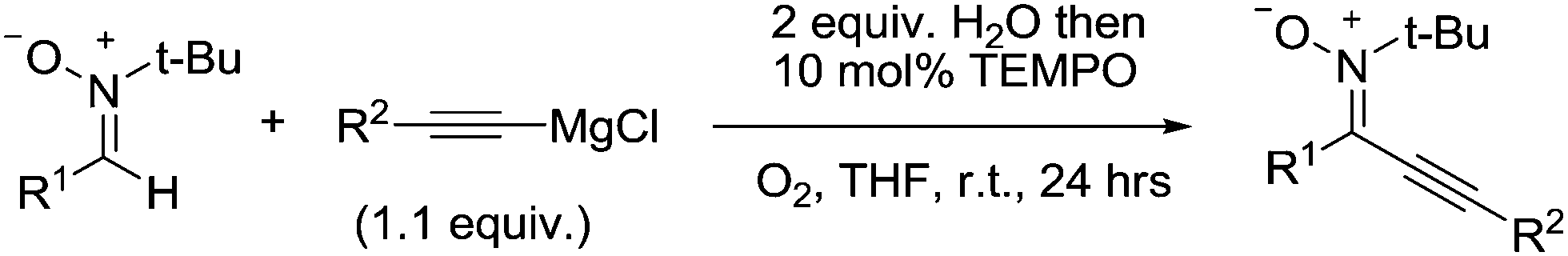 | ||
| Fig. 30 TEMPO mediated cross-coupling reaction between nitrones and alkynyl-Grignard reagents. | ||
This is a useful transformation because the alkynylated nitrone products are valuable precursors for biologically useful isoxazoles. The authors also demonstrated that they could prepare a range of isoxazoles in good yields, using these products.
In 2012, Jiao and co-workers described the use of TEMPO to catalyse the C–C bond formation between two Csp3–H bonds.126 This transformation provided a simple and efficient approach to modify 9,10-dihydroacridine derivatives at the 9-position under the conditions shown in Fig. 31.
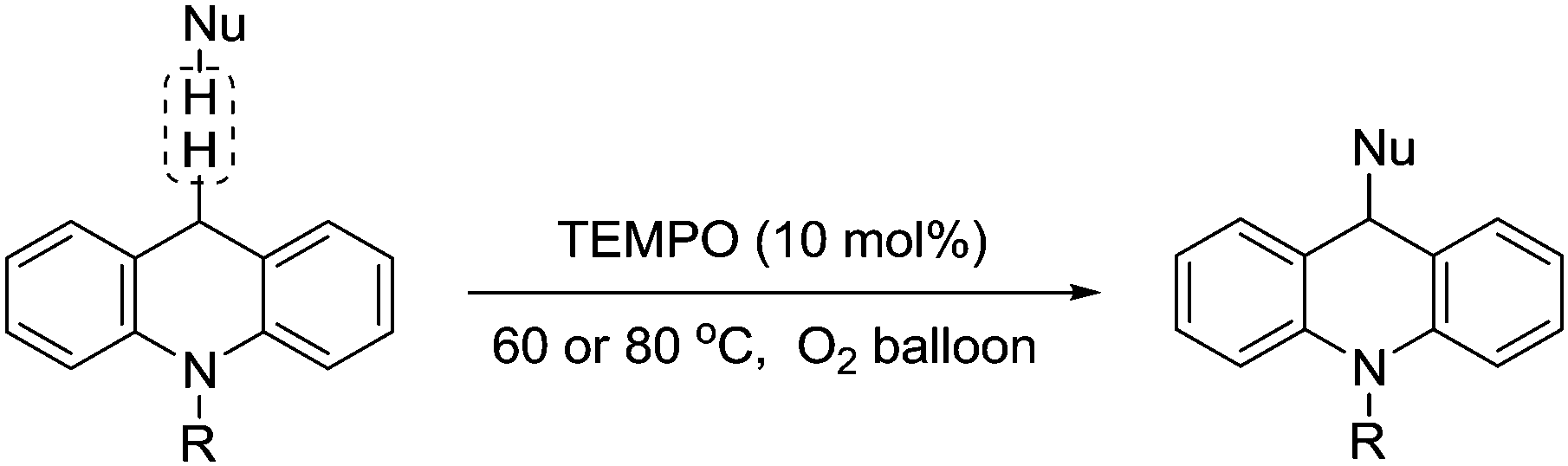 | ||
| Fig. 31 TEMPO-catalysed aerobic oxidative C–C coupling of 9,10-dihydroacridines with carbon nucleophiles. | ||
They demonstrated this for acridines with a range of aliphatic and aromatic groups attached to the nitrogen and a number of different nucleophiles (examples include nitroethane, nitromethane, malonate and malononitrile).
As discussed earlier, Sonobe et al. utilised a Cu/ketoABNO system for the synthesis of imines from amines.105 In this study they also demonstrated a number of C–C bond forming reactions, by reacting the formed imines. They carried out the nucleophilic addition of a Grignard reagent to an imine that had been produced via oxidation catalysis. They also carried out a series of coupling reactions, with some example shown in Fig. 32.
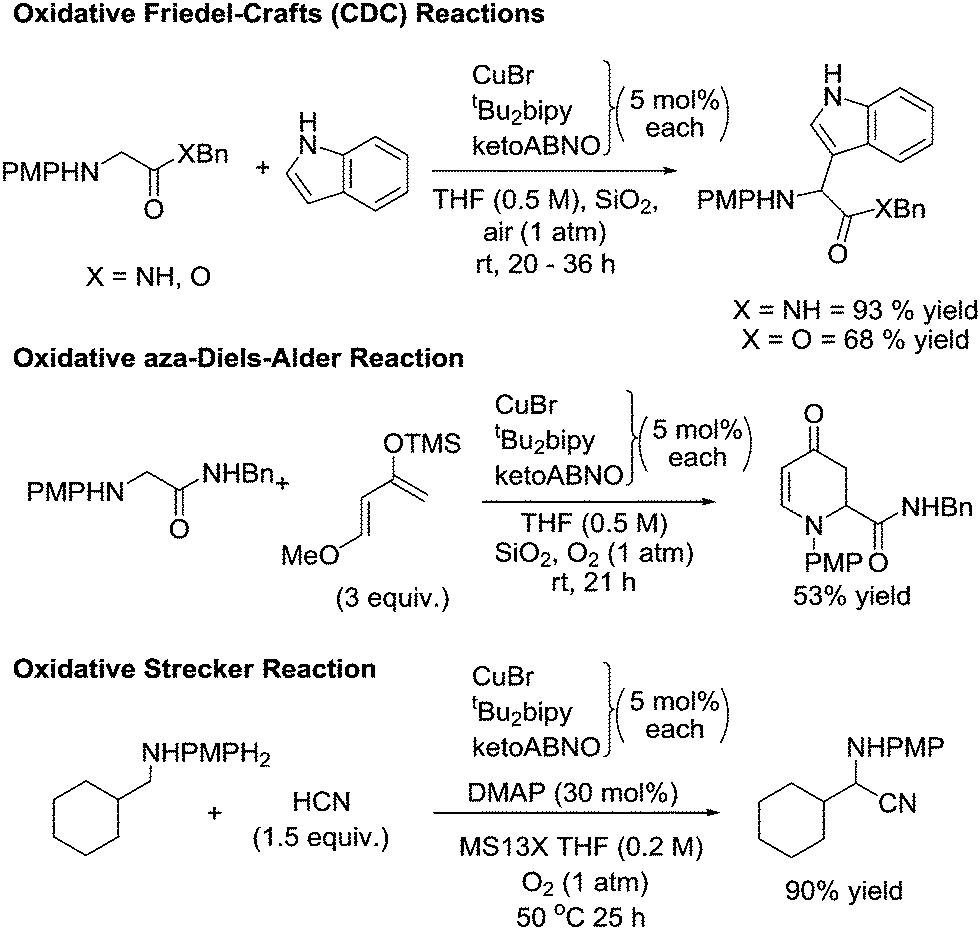 | ||
| Fig. 32 Examples of oxidative reactions of amines using Cu/ketoABNO/O2. | ||
As well as the reactions shown in Fig. 32, they also demonstrated than an asymmetric aerobic CDC reaction could be carried out between a glycine ester and a nitroalkane. There are very few examples of aerobic asymmetric CDC reactions and in this case the copper was ligated with a chiral bis(oxazoline) ligand. This impressive study demonstrates how these oxidation catalysts have the potential to be very valuable tools in organic synthesis.
Indole derivatives are known to exhibit biological activity and are common features of natural products. Liu and co-workers demonstrated that TEMPO could be used as a catalyst for the trimerisation of indoles toward 2,2-disubstituted indolin-3-one derivatives in moderate to excellent yields (Fig. 33).127 In their first study, they utilised TEMPO without any metal co-catalysts, and subsequently they also used CuCl. The use of CuCl enabled the loading of TEMPO to be reduced and in some cases the reaction time was also reduced. The Cu/TEMPO system also delivered better performance for derivatives containing strong electron-withdrawing groups. In the system utilising only TEMPO, when R = NO2 or CN, the desired product was not obtained, while the Cu/TEMPO system afforded these products in moderate yields. They also showed that the Cu/TEMPO system could be used for the dimeric reaction of 2-substituted indoles.
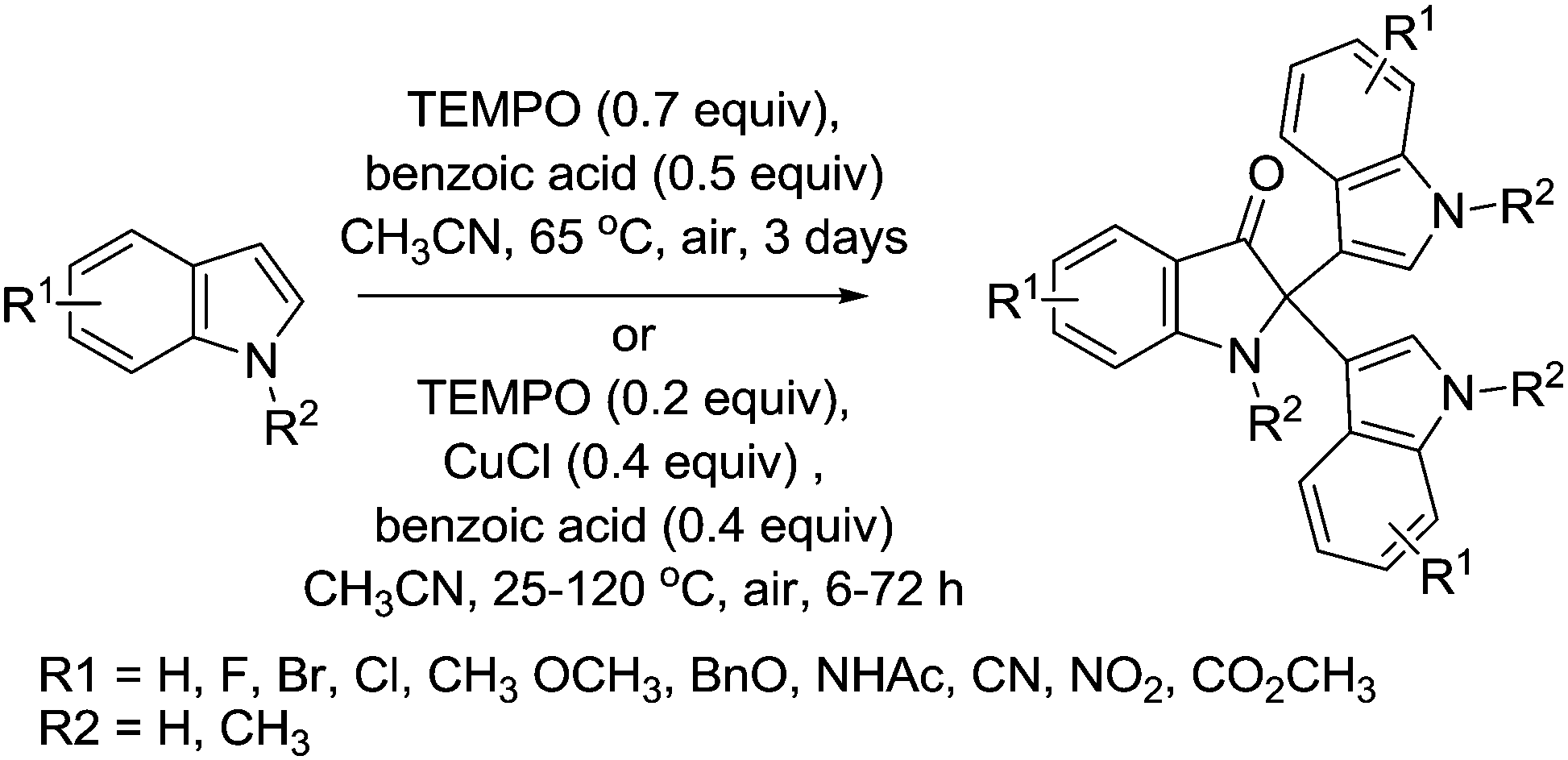 | ||
| Fig. 33 Trimeric reaction of indoles using TEMPO. | ||
Sekar and co-workers reported that TEMPO improved the catalytic performance of copper catalysed enantioselective coupling of 2-napthol derivatives (Fig. 34).128 In this study it was found that the reaction could proceed with just the (R)-BINAM-CuCl catalyst (where BINAM = 1,1′-binaphthyl-2,2′-diamine), but the addition of TEMPO significantly improved the reactivity, enabling these reactions to be carried out at room temperature (rather than 90 °C) in shorter reaction times (2–4 days rather than 7–9 days). A range of 1,1′-binaphthol derivatives were prepared in good to excellent yields (77–98%) with enantioselectivities up to 97% ee (depending on the substituents).
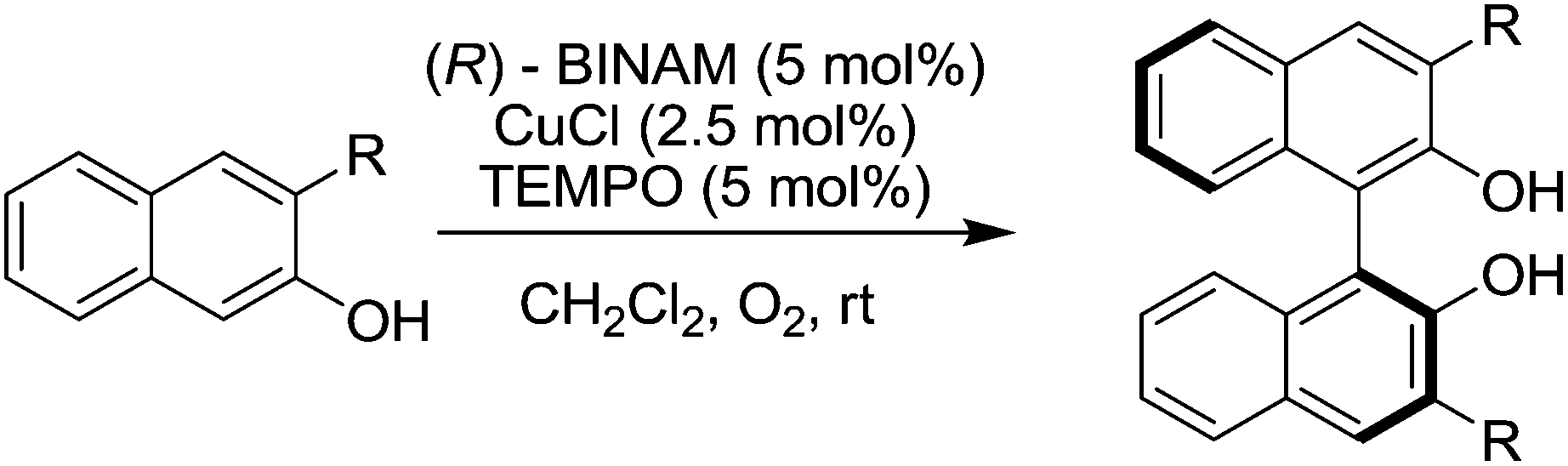 | ||
| Fig. 34 Oxidative coupling of 2-naphthol derivatives. | ||
There are also some recent examples using tris(4-bromophenyl)aminium hexachloroantimonate (TBPA+˙ SbCl6−). This radical cation (Fig. 35) has been used as an aerobic oxidation catalyst in C–C bond forming reactions.
 | ||
| Fig. 35 Tris(4-bromophenyl)aminium hexachloroantimonate. | ||
Jia et al. prepared substituted quinolines from peptides and glycine derivatives using this radical cation.129 Optimisation studies found that in most cases, using InCl3 as a co-catalyst resulted in improved yields and a range of substituted quinolines were prepared with the majority in good yields (Fig. 36).
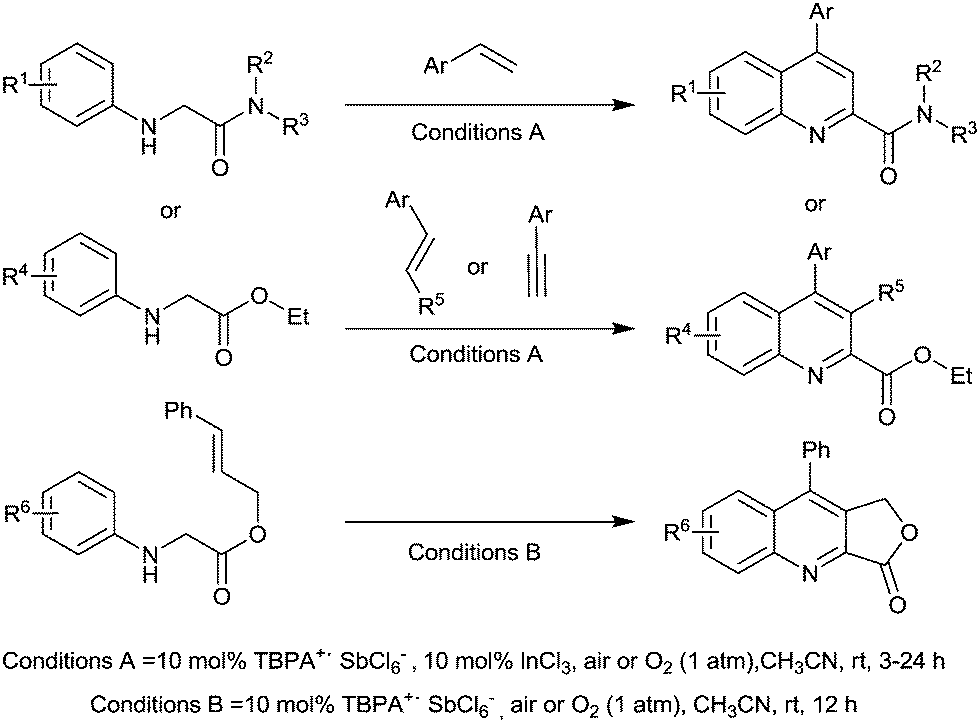 | ||
| Fig. 36 Aerobic synthesis of quinolines using TBPA+˙ SbCl6−. | ||
It was found that O2 was vital for the reaction to take place and the authors proposed some mechanistic details. They believe that it is likely O2 first reacts with TBPA+˙, forming a peroxy radical cation species. This peroxy species is then responsible for initiating the reaction by abstracting a hydrogen atom from the substrate (on the aliphatic carbon adjacent to the nitrogen).
It was shown that TBPA+˙ SbCl6− could be used to catalyse a double Friedel–Crafts reaction of glycine derivatives with indoles (Fig. 37).130 A range of bisindolylmethane derivatives were prepared in good to excellent yields.
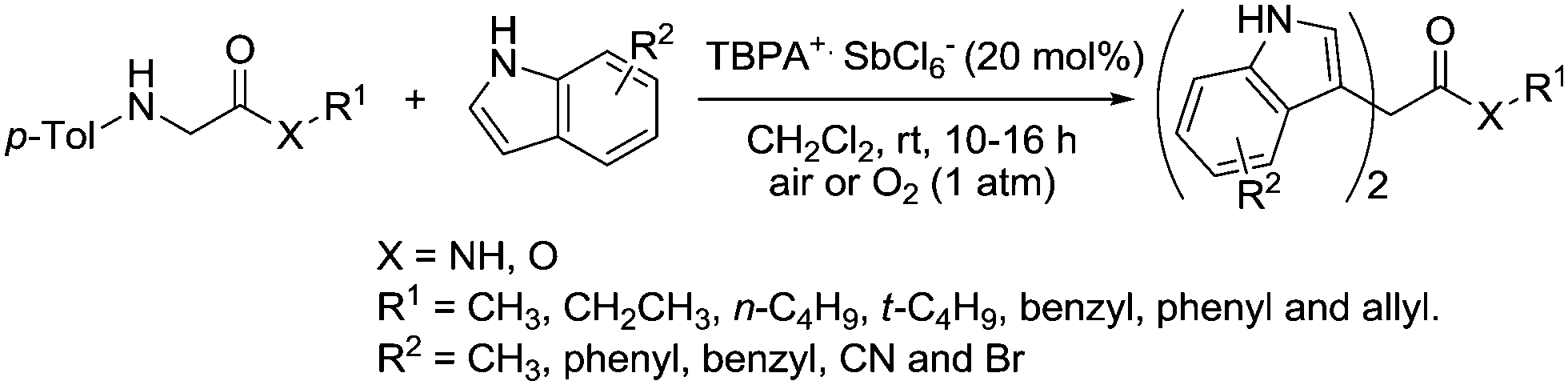 | ||
| Fig. 37 Friedel–Crafts reaction using TBPA+˙ SbCl6−. | ||
The importance of C–C bond formation means that this is likely to be an area that will continue to grow and develop. Further information in this area has been highlighted in a recent review by Mancheño and Stopka which also includes closely related systems not under the remit of this review, for example, non-aerobic systems and the use of oxoammonium salts as stoichiometric oxidants.131
Miscellaneous reactions
In 2011, Yang and co-workers described the use of TEMPO as a catalyst for oxidative S–S bond formation.132 In this example, 2,2′-disbenzothiazole disulphide is used in the production of rubber, however S–S bond formation is generally a useful transformation. It was shown this product could be prepared catalytically using 5 mol% TEMPO (Fig. 38).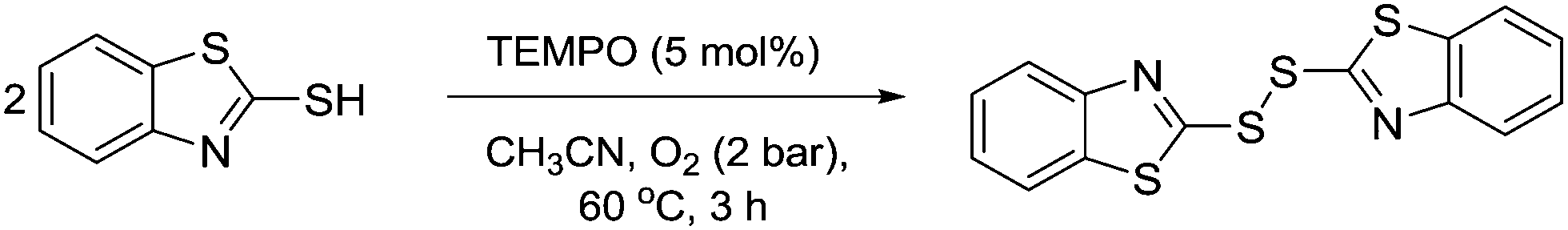 | ||
| Fig. 38 Aerobic oxidative homocoupling of 2-mercap-tobenzothiazole catalysed by TEMPO. | ||
Wang and Jiao discovered that TEMPO could be used as a catalyst for the synthesis of useful oxo nitrile compounds.133 These molecules can be used as precursors for the synthesis of desirable chemicals such as isoquinolines, α-hydroxyketones and alkenenitriles. The TEMPO mediated reaction is outlined in Fig. 39 and involves the cleavage, oxygenation and nitrogenation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. The protocol uses TEMPO as the catalyst and requires the use of trimethylsilyl azide (TMSN3). In the case of benzocycloalkenes, the reaction demonstrated good regioselectivity with oxygenation occurring at the benzylic site. In the case of aliphatic alkenes, the reaction was slower and it was found that the addition of phenyliododiacetate improved the yield. They also looked at terminal alkenes in the form of styrenes, which results in the formation of the corresponding carbonyl compound.
C double bond. The protocol uses TEMPO as the catalyst and requires the use of trimethylsilyl azide (TMSN3). In the case of benzocycloalkenes, the reaction demonstrated good regioselectivity with oxygenation occurring at the benzylic site. In the case of aliphatic alkenes, the reaction was slower and it was found that the addition of phenyliododiacetate improved the yield. They also looked at terminal alkenes in the form of styrenes, which results in the formation of the corresponding carbonyl compound.
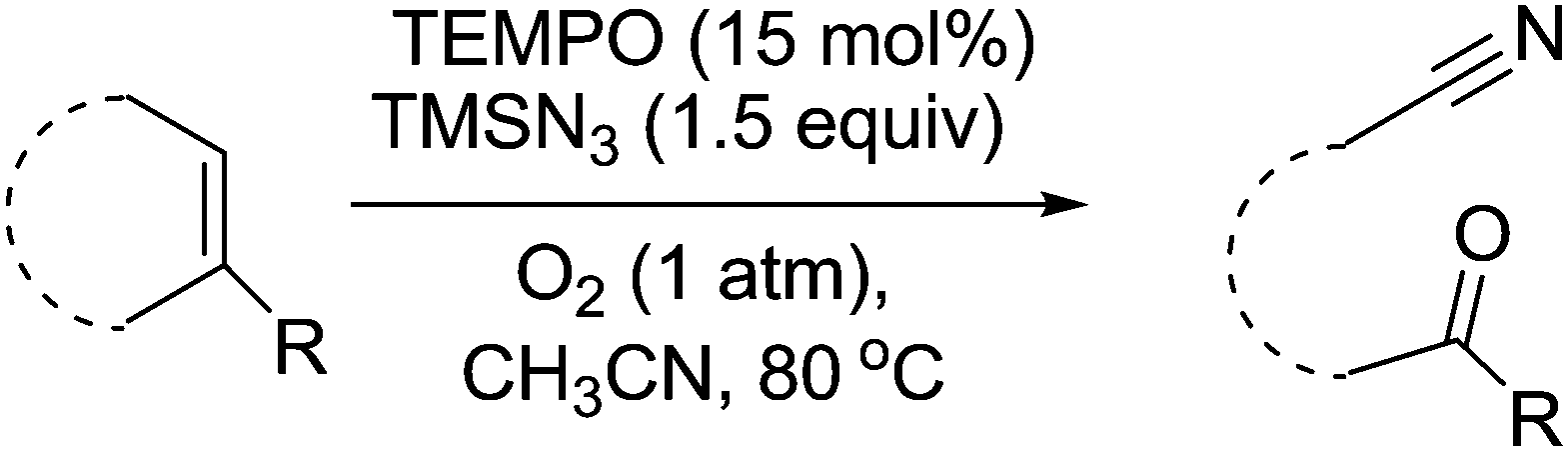 | ||
| Fig. 39 TEMPO mediated cleavage, oxygenation and nitrogenation of CC double bonds to form oxo nitriles. | ||
Ding, Cao and co-workers have described the oxidative deprotection of aldoximes using an FeCl3/TEMPO system (Fig. 40).134 They postulate that cleavage of the oxime produces NO, which then becomes an active species in the catalytic cycle and improves catalytic performance. A number of aldoximes were oxidised to their corresponding aldehydes in excellent isolated yields, however ketoximes were slow to react and resulted in low yields.
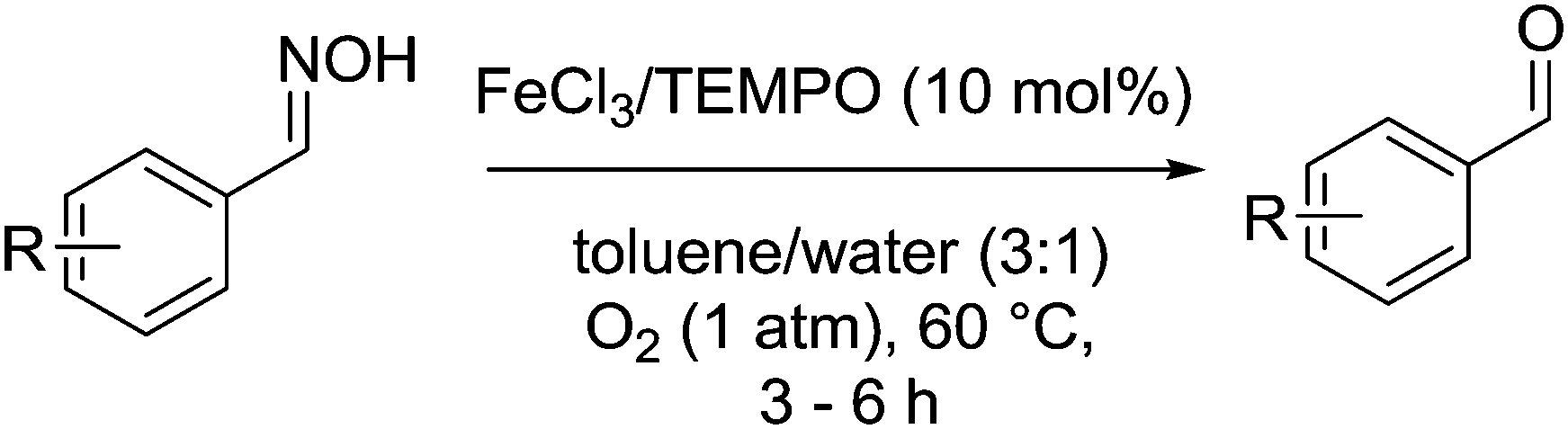 | ||
| Fig. 40 Oxidative deprotection of aldoximes. | ||
Conclusions and outlook
In recent years there have been important strides made in the use of stable radicals for aerobic oxidation catalysis. Alcohol oxidation has gained a lot of research interest and there are now a variety of different methods that can be used efficiently for both primary and secondary alcohols. Importantly, these methods are tolerant of many functionalities and this means that the potential substrate scope is extremely wide. The performance of these systems is arguably at the stage where they could be utilised on a larger industrial scale for the synthesis of some products. There have also been a growing number of reports in which stable radicals are shown to have potential for a wider range of oxidation reactions. In some cases, the performance (e.g. catalyst loading/yields) may not yet be practical, but this is something that should improve as the area matures. We believe that the variety of possible reactions will continue to increase. This may well be driven by a move towards more reactive and sterically diverse radicals. The recent application of radicals such as ABNO and AZADO has already demonstrated that they can have significant advantages over the commonly used TEMPO. The level of sophistication of stable radicals may also increase as it is becoming clear that we can improve the performance of catalysts by tuning both the sterics and electronics of these radicals.76,135,136 As we have highlighted, there are also a few examples where radicals other than nitroxyls have been exploited, and again utilising a wider range of stable radicals may lead to a greater diversity of potential reactions. We believe we are entering an exciting time for this area of chemistry and the prospects for oxidation catalysis with stable radicals are extremely promising.Ultimately the sign of success for a catalytic method is that it is used on a larger, industrial scale. However, we believe there remain a few challenges to such aerobic systems being employed on a larger scale. From a catalyst performance/cost perspective, if these catalysts are to be used for producing less expensive fine chemicals (e.g. fragrance and flavour molecules), then the radicals will need to be recovered and re-used for a process to be economically viable. The cost of radicals such as TEMPO is something that has been previously discussed in relation to industrial applications,5 with the use of solid supported TEMPO highlighted as a way of improving the practicality on a larger scale. Unhindered radicals (e.g. AZADO and ABNO), have emerged as very promising catalysts and these radicals are likely to be more expensive than TEMPO. Consequently, the recovery and re-use of such radicals will be key for some applications.
Another significant obstacle hindering the industrial viability of these catalytic systems is the use of O2 as the terminal oxidant. Although oxygen is utilised on a large scale for the production of commodity chemicals (e.g. terephthalic acid) it is rarely used by the pharmaceutical industry. In a review of large-scale oxidations in the pharmaceutical industry (written by researchers from Pfizer), they stated: “Oxygen gas has the advantage of being the least expensive and most readily available oxidant, but the safety issues surrounding its use make it one of the least frequently used reagents”.137 As mentioned earlier, the majority of industrial applications for TEMPO utilise bleach as the oxidant.5 More recently Holan and Jahn reported the use of TEMPO and AZADO for the oxidation of alcohols.138 In this case, they utilised t-BuONO as a stoichiometric oxidant under anaerobic conditions. The authors point out that such an approach avoids the hazards associated with using O2. In our own opinion, such an approach is probably a more attractive option to those working in the pharmaceutical industry at the present time. But using t-BuONO as a stoichiometric oxidant is most likely not a viable option for the production of cheaper fine chemicals and in these cases aerobic methods would be more desirable.
For aerobic oxidations to be exploited on a larger scale, an important task is demonstrating the use of safer methods for aerobic oxidations. As is evident in this review, it is common in academic studies that pure O2 atmospheres are utilised. On a small scale, the dangers of O2 may be limited but on a larger scale, even air would be too dangerous to use with organic solvents. Utilising air may be viable for aqueous systems (e.g. laccase/nitroxyl systems) but processes using organic solvents reactions require limiting oxygen concentrations (LOC). The LOC is the concentration of oxidant below which combustion is not possible under specified conditions. For most organic solvents (and substrates) an O2 concentration of less than 10% is usually needed to achieve LOC.139 A downside of using dilute O2:N2 gas mixture is that reactions require comparatively higher pressures (e.g. tens of bars) to achieve sufficient O2 concentrations in the liquid phase. Such pressures are another deterrent to those in the pharmaceutical industry. It seems clear that the best way to make such pressurised systems scalable is to use a continuous flow system and as we have highlighted there are already some examples of flow systems with stable radicals. Continuous flow is a standard way of improving the efficiency of a process, but an important aspect of this mode of operation is that it allows much smaller reactors to be used, making the dangers of pressurised systems much easier to handle. The use of microreactors and continuous flow is another area that holds promise as a method of scaling up oxidation reactions. In the case of microreactors it is known that it is possible to work under conditions that would not be suitable using conventional reactor systems; of relevance here is the ability to use pure O2 and work inside the explosive regime without adverse outcomes.140,141 We believe that such approaches to reaction engineering will help aerobic systems become more practical on a larger scale.
Acknowledgements
For support we thank the Department of Employment and Learning, Northern Ireland, Queen's University Belfast Ionic Liquid Laboratories (QUILL) and the Centre for the Theory and Application of Catalysis (CenTACat).References
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 CAS.
- Reviews on catalytic aerobic oxidation reactions: (a) T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037 CrossRef CAS PubMed; (b) S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400 CrossRef CAS PubMed; (c) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329 CrossRef CAS PubMed; (d) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (e) C. P. Vinod, K. Wilson and A. F. Lee, J. Chem. Technol. Biotechnol., 2011, 86, 161 CrossRef CAS; (f) C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547 RSC; (g) E. Roduner, W. Kaim, B. Sarkar, V. B. Urlacher, J. Pleiss, R. Gläser, W.-D. Einicke, G. A. Sprenger, U. Beifuß, E. Klemm, C. Liebner, H. Hieronymus, S.-F. Hsu, B. Plietker and S. Laschat, ChemCatChem, 2013, 5, 82 CrossRef CAS; (h) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed; (i) Book: Modern Oxidation Methods, ed. J.-E. Backvall, Wiley-VCH, Weinheim, 2nd edn, 2010 Search PubMed.
- http://www.bgs.ac.uk/mineralsuk/statistics/riskList.html .
- Guidelines on the specification limits for residues of metal catalysts or metal reagents from the European Agency for the Evaluation of Medicinal Products (EMEA) 2008, (Doc.Ref. EMEA/CHMP/SWP/4446/2000), http://www.emea.europa.eu.
- R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2010, 14, 245 CrossRef CAS.
- L. Anelli, C. Biffi, F. Montanari and S. Quici, J. Org. Chem., 1987, 52, 2559 CrossRef.
- M. M. Zhao, J. Li, E. Mano, Z. J. Song and D. M. Tschaen, Org. Synth., 2005, 81, 195 CrossRef CAS.
- (a) R. A. Sheldon and I. W. C. E. Arends, Adv. Synth. Catal., 2004, 346, 1051 CrossRef CAS; (b) R. A. Sheldon and I. W. C. E. Arends, J. Mol. Catal. A: Chem., 2006, 251, 200 CrossRef CAS.
- J. M. Bobbitt, C. Brückner and N. Merbouh, Org. React., 2009, 74, 106 Search PubMed.
- L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034 CrossRef CAS PubMed.
- S. Wertz and A. Studer, Green Chem., 2013, 15, 3116 RSC.
- Stable Radicals, ed. R. Hicks, John Wiley & Sons, Ltd., Wiltshire, 2010 Search PubMed.
- (a) Y. Ishii, S. Sakaguchi and T. Iwahama, Adv. Synth. Catal., 2001, 343, 393 CrossRef CAS; (b) F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800 CrossRef CAS PubMed; (c) S. Coseri, Catal. Rev.: Sci. Eng., 2009, 51, 218 CrossRef CAS.
- R. M. Dupeyre and A. Rassat, J. Am. Chem. Soc., 1966, 88, 3180 CrossRef CAS.
- J. Bredt, Liebigs Ann. Chem., 1924, 437, 1 CrossRef CAS.
- For example, it was shown that ABNO trapped carbon centered radicals faster than TEMPO: (a) V. W. Bowry and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 4992 CrossRef CAS; (b) A. L. J. Beckwith, V. W. Bowry and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 4983 CrossRef CAS.
- (a) Y. Demizu, H. Shiigi, T. Oda, Y. Matsumura and O. Onomura, Tetrahedron Lett., 2008, 49, 48 CrossRef CAS; (b) H. Shiigi, H. Mori, T. Tanaka, Y. Demizu and O. Onomura, Tetrahedron Lett., 2008, 49, 5247 CrossRef CAS.
- M. Tomizawa, M. Shibuya and Y. Iwabuchi, Org. Lett., 2009, 11, 1829 CrossRef CAS PubMed.
- (a) M. Shibuya, M. Tomizawa, I. Suzuki and Y. Iwabuchi, J. Am. Chem. Soc., 2006, 128, 8412 CrossRef CAS PubMed; (b) M. Shibuya, M. Tomizawa, Y. Sasano and Y. Iwabuchi, J. Org. Chem., 2009, 74, 4619 CrossRef CAS PubMed; (c) M. Shibuya, T. Sato, M. Tomizawa and Y. Iwabuchi, Chem. Commun., 2009, 1739 RSC; (d) M. Hayashi, Y. Sasano, S. Nagasawa, M. Shibuya and Y. Iwabuchi, Chem. Pharm. Bull., 2011, 59, 1570 CrossRef CAS PubMed.
- W. Brackman and C. Gaasbeek, Recl. Trav. Chim. Pays-Bas, 1966, 85, 257 CrossRef CAS.
- (a) M. F. Semmelhack, C. R. Schmid, D. A. Cortes and S. Chou, J. Am. Chem. Soc., 1984, 106, 3374 CrossRef CAS; (b) M. F. Semmelhack, C. R. Schmid and D. A. Cortes, Tetrahedron Lett., 1986, 27, 119 CrossRef.
- (a) N. Jiang and A. J. Ragauskas, ChemSusChem, 2008, 1, 823 CrossRef CAS PubMed; (b) S. Mannam, S. K. Alamsetti and G. Sekar, Adv. Synth. Catal., 2007, 349, 2253 CrossRef CAS; (c) P. J. Figiel, M. Leskelä and T. Repo, Adv. Synth. Catal., 2007, 349, 1173 CrossRef CAS; (d) P. J. Figiel, A. M. Kirillov, Y. Y. Karaback, M. N. Kopylovich and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2009, 305, 178 CrossRef CAS; (e) J. S. Uber, Y. Vogels, D. van den Helder, I. Mutikainen, U. Turpeinen, W. T. Fu, O. Roubeau, P. Gamez and J. Reedijk, Eur. J. Inorg. Chem., 2007, 4197 CrossRef CAS; (f) Z. Lu, J. Sánchez Costa, O. Roebeau, I. Mutikanein, U. Turpeinen, S. J. Teat, P. Gamez and J. Reedijk, Dalton Trans., 2008, 3567 RSC; (g) Z. Lu, T. Ladrak, O. Roubeau, J. van de Toorn, S. J. Teat, C. Massera, P. Gamez and J. Reedijk, Dalton Trans., 2009, 3559 RSC; (h) Q. Wang, Y. Zhang, G. Zheng, Z. Tian and G. Yang, Catal. Commun., 2011, 14, 92 CrossRef CAS; (i) G. Zhang, X. Han, Y. Luan, Y. Wang, X. Wen and C. Ding, Chem. Commun., 2013, 49, 7908 RSC; (j) X. Liu, Q. Xia, Y. Zhang, C. Chen and W. Chen, J. Org. Chem., 2013, 78, 8531 CrossRef CAS PubMed; (k) G. Zhang, X. Hana, Y. Luana, Y. Wanga, X. Wena, L. Xu, C. Dinga and J. Gao, RSC Adv., 2013, 3, 19255 RSC; (l) C. Chen, B. Liu and W. Chen, Synthesis, 2013, 3387 CrossRef CAS; (m) Z. Ma, L. Wei, E. C. B. A. Alegria, L. M. D. R. S. Martins, M. F. C. G. da Silva and A. J. L. Pombeiro, Dalton Trans., 2014, 43, 4048 RSC.
- (a) B. Betzemeir, M. Cavazzin, S. Quici and P. Knochel, Tetrahedron Lett., 2000, 41, 4343 CrossRef; (b) L. Lin, J. Liuyan and W. Yunyang, Catal. Commun., 2008, 9, 1379 CrossRef CAS; (c) L. Lin, M. Juanjuan, J. Liuyan and W. Yunyang, J. Mol. Catal. A: Chem., 2008, 291, 1 CrossRef; (d) M. Herbert, F. Montilla and A. Galindo, Dalton Trans., 2010, 39, 900 RSC; (e) A. Chobrok, S. Baj, W. Pudło and A. Jarzębski, Appl. Catal., A, 2010, 389, 179 CrossRef; (f) N. Lu and Y.-C. Lin, Tetrahedron Lett., 2007, 48, 8823 CrossRef CAS; (g) I. A. Ansari and R. Gree, Org. Lett., 2002, 4, 1507 CrossRef CAS PubMed; (h) N. Jiang and A. J. Ragauskas, Org. Lett., 2005, 7, 3689 CrossRef CAS PubMed.
- (a) L. Liu, J. Ma, J. Xia, L. Lia, C. Lia, X. Zhanga, J. Gonga and Z. Tong, Catal. Commun., 2011, 12, 323 CrossRef CAS; (b) L. Liu, D. Liu, Z. Xia, J. Gao, T. Zhang, J. Ma, D. Zhang and Z. Tong, Monatsh. Chem., 2013, 144, 251 CrossRef CAS; (c) S. Samanta, S. Das, P. K. Samanta, S. Dutta and P. Biswas, RSC Adv., 2013, 3, 19455 RSC.
- N. Mase, T. Mizumori and Y. Tatemoto, Chem. Commun., 2011, 47, 2086 RSC.
- T. S. Hansen, I. Sádaba, E. J. García-Suárez and A. Riisager, Appl. Catal., A, 2013, 456, 44 CrossRef CAS.
- (a) P. Gamez, I. W. C. E. Arends, J. Reedijk and R. A. Sheldon, Chem. Commun., 2003, 2414 RSC; (b) A. Dijksman, I. W. C. E. Arends and R. A. Sheldon, Org. Biomol. Chem., 2003, 1, 3232 RSC; (c) P. Gamez, I. W. C. E. Arends, R. A. Sheldon and J. Reedijk, Adv. Synth. Catal., 2004, 346, 805 CrossRef CAS.
- (a) J. W. Whittaker, Chem. Rev., 2003, 103, 2347 CrossRef CAS PubMed; (b) L. Que and W. B. Tolman, Nature, 2008, 455, 333 CrossRef CAS PubMed.
- E. T. T. Kumpulainen and A. M. P. Koskinen, Chem. – Eur. J., 2009, 15, 10901 CrossRef CAS PubMed.
- J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901 CrossRef CAS PubMed.
- J. M. Hoover, J. E. Steves and S. S. Stahl, Nat. Protoc., 2012, 7, 1161 CrossRef CAS PubMed.
- N. J. Hill, J. M. Hoover and S. S. Stahl, J. Chem. Educ., 2013, 90, 102 CrossRef CAS.
- J. F. Greene, J. M Hoover, D. S. Mannel, T. W. Root and S. S. Stahl, Org. Process Res. Dev., 2013, 17, 1247 CrossRef CAS.
- J. M Hoover, B. L. Ryland and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 23 CrossRef PubMed.
- J. M. Hoover, B. L. Ryland and S. S. Stahl, ACS Catal., 2013, 3, 2599 CrossRef CAS PubMed.
- I. Gamba, I. Mutikainen, E. Bouwman, J. Reedijk and S. Bonnet, Eur. J. Inorg. Chem., 2013, 115 CrossRef CAS.
- P. Belanzoni, C. Michel and E. J. Baerends, Inorg. Chem., 2011, 50, 11896 CrossRef CAS PubMed.
- C. Michel, P. Belanzoni, P. Gamez, J. Reedijk and E. J. Baerans, Inorg. Chem., 2009, 48, 11909 CrossRef CAS PubMed.
- L. Cheng, J. Wang, M. Wang and Z. Wu, Inorg. Chem., 2010, 49, 9392 CrossRef CAS PubMed.
- M. M. Hossain and S.-G. Shyu, Adv. Synth. Catal., 2010, 352, 3061 CrossRef CAS.
- J. E. Steves and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 15742 CrossRef CAS PubMed.
- Y. Sasano, S. Nagasawa, M. Yamazaki, M. Shibuya, J. Park and Y. Iwabuchi, Angew. Chem., Int. Ed., 2014, 53, 3236 CrossRef CAS PubMed.
- N. Wang, R. Liu, J. Chen and X. Liang, Chem. Commun., 2005, 5322 RSC.
- X. Wang and X. Liang, Chin. J. Catal., 2008, 29, 935 CrossRef CAS.
- W. Yin, C. Chu, Q. Lu, J. Tao, X. Liang and R. Liu, Adv. Synth. Catal., 2010, 352, 113 CrossRef CAS.
- C.-X. Miao, J.-Q. Wang, B. Yu, W.-G. Cheng, J. Sun, S. Chanfreau, L.-N. He and S.-J. Zhang, Chem. Commun., 2011, 47, 2697 RSC.
- S. Ma, J. Liu, S. Li, B. Chen, J. Cheng, J. Kuang, Y. Liu, B. Wan, Y. Wang, J. Ye, Q. Yu, W. Yuan and S. Yu, Adv. Synth. Catal., 2011, 353, 1005 CrossRef CAS.
- J. J. Scepaniak, A. M. Wright, R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 19350 CrossRef CAS PubMed.
- (a) S. Chou, J. A. Nelson and T. A. Spencer, J. Org. Chem., 1974, 39, 2356 CrossRef; (b) Z. Ma, Q. Huang and J. M. Bobbitt, J. Org. Chem., 1993, 58, 4837 CrossRef CAS.
- J. Liu and S. Ma, Org. Biomol. Chem., 2013, 11, 4186 CAS.
- (a) J. Liu and S. Ma, Synthesis, 2013, 1624 CAS; (b) J. Liu and S. Ma, Tetrahedron, 2013, 69, 10161 CrossRef CAS.
- J. Liu and S. Ma, Org. Lett., 2013, 15, 5150 CrossRef CAS PubMed.
- L. Wang, J. Li, Y. Lv, G. Zhao and S. Gao, Appl. Organomet. Chem., 2012, 26, 37 CrossRef CAS.
- L. Wang, J. Li, X. Zhao, Y. Lv, H. Zhang and S. Gao, Tetrahedron, 2013, 69, 6041 CrossRef CAS.
- (a) C. Bolm and T. Fey, Chem. Commun., 1999, 1795 RSC; (b) C. Bolm, A. S. Magnus and J. P. Hildebrand, Org. Lett., 2000, 2, 1173 CrossRef CAS PubMed.
- D. Obermayer, A. M. Balu, A. A. Romero, W. Goessler, R. Luque and C. O. Kappe, Green Chem., 2013, 15, 1530 RSC.
- A. Cecchetto, F. Fontana, F. Minisci and F. Recupero, Tetrahedron Lett., 2001, 42, 6651 CrossRef CAS.
- F. Minisci, F. Recupero, M. Rodinò, M. Sala and A. Schneider, Org. Process Res. Dev., 2003, 7, 794 CrossRef CAS.
- F. Minisci, F. Recupero, A. Cecchetto, C. Gambarotti, C. Punta, R. Faletti, R. Paganelli and G. F. Pedulli, Eur. J. Org. Chem., 2004, 109 CrossRef CAS.
- M. Gilhespy, M. Lok and X. Baucherel, Chem. Commun., 2005, 1085 RSC.
- M. Gilhespy, M. Lok and X. Baucherel, Catal. Today, 2006, 117, 114 CrossRef CAS.
- G. Yang, J. Ma, W. Wang, J. Zhao, X. Lin, L. Zhou and X. Gao, Catal. Lett., 2006, 112, 83 CrossRef CAS.
- Y. Guo, J. Zhao, J. Xu, W. Wang, F. Tian, G. Yang and M. Song, J. Nat. Gas Chem., 2007, 16, 210 CrossRef CAS.
- G. Yang, W. Zhu, P. Zhang, H. Xue, W. Wang, J. Tian and M. Song, Adv. Synth. Catal., 2008, 350, 542 CrossRef CAS.
- Y. Jing, J. Jiang, B. Yan, S. Lu, J. Jiao, H. Xue, G. Yang and G. Zheng, Adv. Synth. Catal., 2011, 353, 1146 CrossRef CAS.
- R. Ben-Daniel, P. Alsters and R. Neumann, J. Org. Chem., 2001, 66, 8650 CrossRef CAS PubMed.
- J. Zhu, P.-C. Weng and M. Lu, RSC Adv., 2012, 2, 8265 RSC.
- Z. Du, J. Ma, H. Ma, M. Wang, Y. Huang and J. Xu, Catal. Commun., 2010, 11, 732 CrossRef CAS.
- M. Fabbrini, C. Galli, P. Gentili and D. Machitella, Tetrahedron Lett., 2001, 42, 7551 CrossRef CAS.
- (a) F. D'Acunzo, P. Baiocco, M. Fabbrini, C. Galli and P. Gentili, Eur. J. Org. Chem., 2002, 4195 CrossRef; (b) C. Galli and P. Gentilli, J. Phys. Org. Chem., 2004, 17, 973 CrossRef CAS; (c) P. Astolfi, P. Brandi, C. Galli, P. Gentili, M. F. Gerini, L. Greci and O. Lanzalunga, New J. Chem., 2005, 29, 1308 RSC; (d) S. A Tromp, I. Matijošytė, R. A. Sheldon, I. W. C. E. Arends, G. Mul, M. T. Kreutzer, J. A Moulijn and S. de Vries, ChemCatChem, 2010, 2, 827 CrossRef.
- S. R. Couto and J. L. T. Herrera, Biotechnol. Adv., 2006, 24, 500 CrossRef PubMed.
- S. Witayakran and A. J. Ragauskas, Adv. Synth. Catal., 2009, 351, 1187 CrossRef CAS.
- C. Zhu, Z. Zhang, W. Ding, J. Xie, Y. Chen, J. Wu, X. Chen and H. Ying, Green Chem., 2014, 16, 1131 RSC.
- (a) R. Liu, X. Liang, C. Dong and X. Hu, J. Am. Chem. Soc., 2004, 126, 4112 CrossRef CAS PubMed; (b) R. Liu, C. Dong, X. Liang, X. Wang and X. Hu, J. Org. Chem., 2005, 70, 729 CrossRef CAS PubMed; (c) X. Wang, R. Liu, Y. Jin and X. Liang, Chem. – Eur. J., 2008, 14, 2679 CrossRef CAS PubMed; (d) J. Zhang, Z. Jiang, D. Zhao, G. He, S. Zhou and S. Han, Chin. J. Chem., 2013, 31, 794 CrossRef CAS; (e) C. X. Miao, L. N. He, J. Q. Wang and J. L. Wang, Adv. Synth. Catal., 2009, 351, 2209 CrossRef CAS; (f) C. X. Miao, L. N. He, J. L. Wang and F. Wu, J. Org. Chem., 2010, 75, 257 CrossRef CAS PubMed; (g) Y. Xie, W. Mo, D. Xu, Z. Shen, N. Sun, B. Hu and X. Hu, J. Org. Chem., 2007, 72, 4288 CrossRef CAS PubMed; (h) G. Yang, W. Wang, W. Zhu, C. An, X. Gao and M. Song, Synlett, 2010, 437 CrossRef CAS; (i) Y. Xie, W. Mo, D. Xu, Z. Shen, N. Sun, B. Hu and X. Hu, J. Org. Chem., 2007, 72, 4288 CrossRef CAS PubMed.
- For example: (a) X. He, Z. Shen, W. Mo, N. Sun, B. Hu and X. Hu, Adv. Synth. Catal., 2009, 351, 89 CrossRef CAS; (b) Y. Kuang, H. Rokubuichi, Y. Nabae, T. Hayakawa and M. Kakimoto, Adv. Synth. Catal., 2010, 352, 2635 CrossRef CAS; (c) S. Wertz and A. Studer, Adv. Synth. Catal., 2011, 353, 69 CrossRef CAS.
- M. Shibuya, Y. Osada, Y. Sasano, M. Tomizawa and Y. Iwabuchi, J. Am. Chem. Soc., 2011, 133, 6497 CrossRef CAS PubMed.
- Y. Kuang, Y. Nabae, T. Hayakawa and M. Kakimoto, Green Chem., 2011, 13, 1659 RSC.
- M. B. Lauber and S. S. Stahl, ACS Catal., 2013, 3, 2612 CrossRef CAS.
- J. Zhu, P.-C. Wang and L. Ming, Synth. Commun., 2013, 43, 1871 CrossRef CAS.
- R. Prebil, G. Stavber and S. Stavber, Eur. J. Org. Chem., 2014, 395 CrossRef CAS.
- B. Karimi, A. Biglari, J. H. Clarke and V. Budarin, Angew. Chem., Int. Ed., 2007, 46, 7210 CrossRef CAS PubMed.
- B. Karimi and E. Badreh, Org. Biomol. Chem., 2011, 9, 4194 CAS.
- B. Karimi and E. Farhangi, Chem. – Eur. J., 2011, 17, 6056 CrossRef CAS PubMed.
- (a) L. Di and Z. Hua, Adv. Synth. Catal., 2011, 353, 1253 CrossRef CAS; (b) H. Zhang, L. Fu and H. Zhong, Chin. J. Catal., 2013, 34, 1848 CrossRef CAS; (c) H. Zhang and L. Fu, Synth. Commun., 2014, 44, 610 CrossRef CAS.
- P. Ionita, RSC Adv., 2013, 3, 21218 RSC.
- C. Aellig, D. Scholz, S. Conrad and I. Hermans, Green Chem., 2013, 15, 1975 RSC.
- D. Könning, W. Hiller and M. Christmann, Org. Lett., 2012, 14(20), 5258 CrossRef PubMed.
- X.-H. Ho, H.-J. Oh and H.-Y. Jang, Eur. J. Org. Chem., 2012, 5655 CrossRef CAS.
- (a) J. Brioche, G. Masson and J. Zhu, Org. Lett., 2010, 12, 1432 CrossRef CAS PubMed; (b) B. Karimi and E. Farhangi, Adv. Synth. Catal., 2013, 355, 508 CAS.
- (a) B. Sedai, C. Díaz-Urrutia, R. T. Baker, R. Wu, L. A. Silks and S. K. Hanson, ACS Catal., 2011, 1, 794 CrossRef CAS; (b) B. Sedai, C. Díaz-Urrutia, R. T. Baker, R. Wu, L. A. Silks and S. K. Hanson, ACS Catal., 2013, 3, 3111 CrossRef CAS.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415 CrossRef CAS PubMed.
- C. Zhang and N. Jiao, J. Am. Chem. Soc., 2010, 132, 28 CrossRef CAS PubMed.
- S. C. Ghosh, J. S. Y. Ngiam, A. M. Seayad, D. T. Tuan, C. W. Johannes and A. Chen, Tetrahedron Lett., 2013, 54, 4922 CrossRef CAS.
- H. Zhao, M. Wang, W. Su and M. Hong, Adv. Synth. Catal., 2010, 352, 1301 CrossRef CAS.
- S. Maity, S. Manna, S. Rana, T. Naveen, A. Mallick and D. Maiti, J. Am. Chem. Soc., 2013, 135, 3355 CrossRef CAS PubMed.
- T. Naveen, S. Maity, U. Sharma and D. Maiti, J. Org. Chem., 2013, 78, 5949 CrossRef CAS PubMed.
- S. Maity, T. Naveen, U. Sharma and D. Maiti, Org. Lett., 2013, 15, 3384 CrossRef CAS PubMed.
- S. Manna, S. Jana, T. Saboo, A. Maji and D. Maiti, Chem. Commun., 2013, 49, 5286 RSC.
- Recent microreview on catalytic oxidation of amines to imines: M. Largeron, Eur. J. Org. Chem., 2013, 5225 CrossRef CAS.
- L. Jiang, L. Jin, H. Tian, X. Yuan, X. Yu and Q. Xu, Chem. Commun., 2011, 47, 10833 RSC.
- H. Tian, X. Yu, Q. Li, J. Wang and Q. Xu, Adv. Synth. Catal., 2012, 354, 2671 CrossRef CAS.
- Z. Hu and F. M. Kerton, Org. Biomol. Chem., 2012, 10, 1618 CAS.
- B. Huang, H. Tian, S. Lin, M. Xie, X. Yu and Q. Xu, Tetrahedron Lett., 2013, 54, 2861 CrossRef CAS.
- E. Zhang, H. Tian, S. Xu, X. Yu and Q. Xu, Org. Lett., 2013, 15, 2704 CrossRef CAS PubMed.
- T. Sonobe, K. Oisaki and M. Kanai, Chem. Sci., 2012, 3, 3249 RSC.
- S. Wertz and A. Studer, Helv. Chim. Acta, 2012, 95, 1758 CrossRef CAS.
- (a) K. Suzuki, T. Watanabe and S.-I. Murahashi, Angew. Chem., Int. Ed., 2008, 47, 2079 CrossRef CAS PubMed; (b) K. Suzuki, T. Watanabe and S.-I. Murahashi, J. Org. Chem., 2013, 78, 2301 CrossRef CAS PubMed.
- Y.-X. Chen, L.-F. Qian, W. Zhang and B. Han, Angew. Chem., Int. Ed., 2008, 47, 9330 CrossRef CAS PubMed.
- B. Han, C. Wang, R.-F. Han, W. Yu, X.-Y. Duan, R. Fang and X.-L. Yang, Chem. Commun., 2011, 47, 7818 RSC.
- B. Han, X.-L. Yang, C. Wang, Y.-W. Bai, T.-C. Pan, X. Chen and W. Yu, J. Org. Chem., 2012, 77, 1136 CrossRef CAS PubMed.
- Z. Chen, J. Chen, M. Liu, J. Ding, W. Cao, X. Huang and H. Wu, J. Org. Chem., 2013, 78, 11342 CrossRef CAS PubMed.
- J. C. A. Flanagan, L. M. Dornan, M. G. McLaughlin, N. G. McCreanor, M. J. Cook and M. J. Muldoon, Green Chem., 2012, 14, 1281 RSC.
- B.-H. Lou, S.-B. Chen, J. Wang, Y. Chen and J.-H. Li, J. Chem. Res., 2013, 37, 409 CrossRef CAS.
- H. Richter, R. Fröhlich, C.-G. Daniliuc and O. G. Mancheño, Angew. Chem., Int. Ed., 2012, 51, 8656 CrossRef CAS PubMed.
- H. Richter and O. G. Mancheño, Org. Lett., 2011, 13, 6066 CrossRef CAS PubMed.
- R. Rohlmann, T. Stopka, H. Richter and O. G. Mancheño, J. Org. Chem., 2013, 78, 6050 CrossRef CAS PubMed.
- X. Zhu, Y.-F. Wang, W. Ren, F.-L. Zhang and S. Chiba, Org. Lett., 2013, 15, 3214 CrossRef CAS PubMed.
- C. Tao, F. Liu, Y. Zhu, W. Liu and Z. Cao, Org. Biomol. Chem., 2013, 11, 3349 CAS.
- W. Yin, C. Wang and Y. Huang, Org. Lett., 2013, 15, 1850 CrossRef CAS PubMed.
- L. M. Dornan, Q. Cao, J. C. A. Flanagan, J. J. Crawford, M. J. Cook and M. J. Muldoon, Chem. Commun., 2013, 49, 6030 RSC.
- J. Kim and S. S. Stahl, ACS Catal., 2013, 3, 1652 CrossRef CAS PubMed.
- M. F. Semmelhack and C. R. Schmid, J. Am. Chem. Soc., 1983, 105, 6732 CrossRef CAS.
- Review on aerobic oxidation of amines: M. T. Schümperli, C. Hammond and I. Hermans, ACS Catal., 2012, 2, 1108 CrossRef.
- M. S. Maji, T. Pfeifer and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 9547 CrossRef CAS PubMed.
- S. Murarka and A. Studer, Adv. Synth. Catal., 2011, 353, 2708 CrossRef CAS.
- B. Zhang, Y. Cui and N. Jiao, Chem. Commun., 2012, 48, 4498 RSC.
- (a) W.-B. Qin, Q. Chang, Y.-H. Bao, N. Wang, Z.-W. Chen and L.-X. Liu, Org. Biomol. Chem., 2012, 10, 8814 RSC; (b) Y.-B. Kong, J.-Y. Zhu, Z.-W. Chen and L.-X. Liu, Can. J. Chem., 2014, 92, 269 CrossRef CAS.
- S. K. Alamsetti, E. Poonguzhali, D. Ganapathy and G. Sekar, Adv. Synth. Catal., 2013, 355, 2803 CrossRef CAS.
- (a) X. Jia, F. Peng, C. Qing, C. Huo and X. Wang, Org. Lett., 2012, 14, 4030 CrossRef CAS PubMed; (b) X. Jia, Y. Wang, F. Peng, C. Huo, L. Yu, J. Liu and X. Wang, J. Org. Chem., 2013, 78, 9450 CrossRef CAS PubMed.
- C. Huo, C. Wang, C. Sun, X. Jia, X. Wang, W. Chang and M. Wu, Adv. Synth. Catal., 2013, 355, 1911 CrossRef CAS.
- O. G. Mancheño and T. Stopka, Synthesis, 2013, 1602 CrossRef.
- B. Dong, S. Lu, J. Jiang, W. Zhang, W. Zhu, M. Tang and G. Yang, J. Chem. Technol. Biotechnol., 2012, 87, 341 CrossRef CAS.
- T. Wang and N. Jiao, J. Am. Chem. Soc., 2013, 135, 11692 CrossRef CAS PubMed.
- G. Zhang, X. Wen, Y. Wang, X. Han, Y. Luan, L. Zheng, C. Ding and X. Cao, RSC Adv., 2013, 3, 22918 RSC.
- A recent example of a nitroxyl radicals with electron-withdrawing ester groups (although not used aerobically in this case): S. Hamada, T. Furuta and Y. Wada, Angew. Chem., Int. Ed., 2013, 52, 8093 CrossRef CAS PubMed.
- J. A. Bogart, H. B. Lee, M. A. Boreen, M. Jun and E. J. Schelter, J. Org. Chem., 2013, 78, 6344 CrossRef CAS PubMed.
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. Brown Ripin, Chem. Rev., 2006, 106, 2943 CrossRef CAS PubMed.
- M. Holan and U. Jahn, Org. Lett., 2014, 16, 58 CrossRef CAS PubMed.
- E. Brandes and W. Möller, Safety Characteristic Data: Flammable Liquids and Gases, Wirtschaftsverlag NW, Bremerhaven, 2008, vol. 1 Search PubMed.
- Review: K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406 CrossRef PubMed.
- Examples of oxidations using O2 in the explosive regime: (a) H. Ge, G. Chen, Q. Yuan and H. Li, Catal. Today, 2005, 110, 171 CrossRef CAS; (b) J. Fischer, T. Lange, R. Boehling, A. Rehfinger and E. Klemm, Chem. Eng. Sci., 2010, 65, 4866 CrossRef CAS; (c) T. Lange, S. Heinrich, C. Liebner, H. Hieronymus and E. Klemm, Chem. Eng. Sci., 2012, 69, 440 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |