 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed cyclization of bromoenynamides to tricyclic azacycles: synthesis of trikentrin-like frameworks†
Craig D.
Campbell
a,
Rebecca L.
Greenaway
a,
Oliver T.
Holton
a,
Helen A.
Chapman
b and
Edward A.
Anderson
*a
aChemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: edward.anderson@chem.ox.ac.uk; Fax: +44 (0)1865 285002; Tel: +44 (0)1865 285000
bSyngenta Ltd., Jealott's Hill International Research Centre, Bracknell, Berkshire RG42 6EY, UK
First published on 22nd August 2013
Abstract
Palladium-catalyzed cascade cyclization of bromoenynamides equipped with an additional alkyne or ynamide substituent affords azatricyclic products. Using 5- to 7-membered ring tethers, this chemistry offers a regiospecific route to highly-functionalized azacycles. Elaboration to the trikentrin B skeleton is achieved from the arylsilane cyclization products.
The trikentrin and herbindole families of natural products, isolated from the marine sponges Trikentrion flabelliforme1a and Axinella sp.,1b display a range of bioactivities including antimicrobial, antifeedant and cytotoxic properties. The heavily-substituted tricyclic indole systems which feature in these compounds (e.g. cis-trikentrin B and herbindole B, Scheme 1) represent a particular synthetic challenge that has inspired a number of elegant solutions.1
 | ||
| Scheme 1 cis-Trikentrin B, herbindole B, and the general bromoenynamide cyclization strategy. | ||
In recent work, we have developed a number of routes to azabicycles from ynamides, including viaynamide carbopalladation.2 We have also reported a strategy to access the tricyclic 7,6,5-CDE ring cores of rubriflordilactones A and B, which contain penta- and tetrasubstituted arenes respectively, through palladium-catalyzed cascade cyclization of bromoenediynes.3 We noted that the combination of these two methodologies could provide access to the tricyclic indole core of the trikentrins, viacyclization of a bromoenynamide equipped with a remote alkyne (1 → 2, Scheme 1). The construction of fused ring arenes in this manner4 has rarely been employed in synthetic endeavours,3 and in the context of ynamides offers a useful alternative to the elegant cyclotrimerization methodology pioneered by Witulski,5 which has recently been applied to the herbindole system.1c This sequenced carbopalladation strategy offers advantages over intramolecular cyclotrimerization, where long tethers restrict the formation of larger rings, presumably due to competing intermolecular reactions.6 Here we describe the development of this novel ynamide chemistry,7 and its application to a number of azatricyclic systems, including aza- and benzazepine trikentrin analogues. Elaboration to the bis-desmethyl-trikentrin B framework is also described.
We first set about the synthesis of a series of bromoenynamide alkynes suitable for cyclization, through the preparation of appropriate sulfonamide and bromoalkyne precursors (Scheme 2). The bromoalkenyl sulfonamides 3a and 3b were prepared from alkynes4a and 4bvia bromoboration/protodeborylation (with simultaneous Boc deprotection), whilst bromoalkynes 5a and 5b were synthesized from 1,6-heptadiyne by monosilylation then bromination. Both trimethylsilyl and benzyldimethylsilyl groups were installed, which we anticipated would enable various strategies for the attachment of trikentrin-like sidechains following cyclization. These building blocks were coupled using Hsung's copper-catalyzed methodology for ynamide formation,8 which provided ynamides 1a–c in moderate to good yield, albeit accompanied by a degree of desilylation in the case of TMS-substituted diyne 5a. To further extend the cyclization methodology, we targeted a substrate featuring two ynamides, which would lead to an aza-trikentrin (pyrroloindoline) framework. The bis-ynamide 1d was readily prepared in four steps from 4a by bromination/carbamate deprotection (to afford the sulfonamide6), followed by sequential Hsung couplings – firstly with 1-bromo-oct-1-yne (used in excess to minimize intermolecular homocoupling of 6), then with sulfonamide3a.
 | ||
| Scheme 2 Preparation of ynamide and bis-ynamide cyclization substrates. | ||
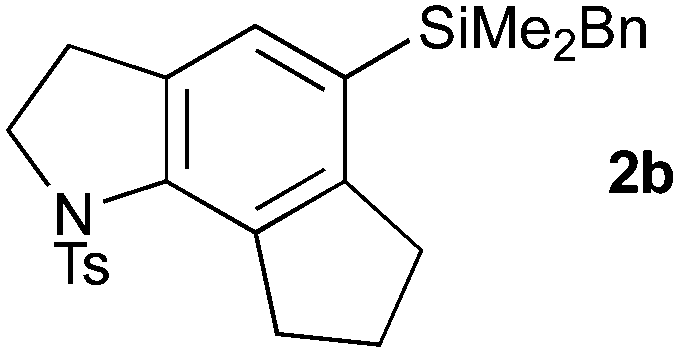
With a selection of substrates in hand, the cascade cyclizations were investigated (Table 1). Using our previously reported conditions (10 mol% Pd(PPh3)4, Et3N, 0.017 M in MeCN, 80 °C),3 we were pleased to obtain the 5,6,5-tricyclic trikentrin frameworks 2a and 2b in excellent yields (90%, entries 1 and 4). The catalyst loading could be lowered to 5 mol% with a slight reduction in yield (entries 2 and 5); however, by increasing the concentration (to 0.16 M), catalytic efficiency was restored, with 2a isolated in 88% yield (entry 3).


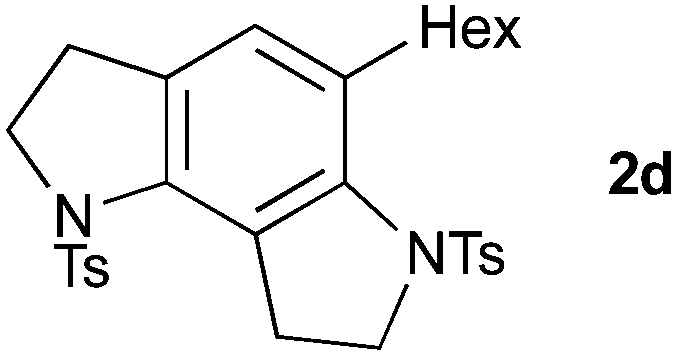
Cyclization to the challenging 7,6,5-tricyclic analogue of the trikentrin framework was next attempted. At higher catalyst loading and dilution, the desired tricycle 2c was obtained as a component of a complex mixture (entry 6). However, by performing this reaction at higher concentration, 2c was formed as the sole product in excellent yield (entry 7), a result that highlights the advantages of the sequenced carbopalladation strategy. Finally, diynamide 1d was subjected to the range of reaction conditions (entries 8–10). To our delight, tricycle 2d, which represents the first example of such a pyrroloindoline framework, was isolated in high yield when reacted at the higher concentration (70%, entry 10).
With efficient access to azatricycles established, we aimed to demonstrate the utility of the methodology by preparing a natural product analogue – bis-desmethyl-trikentrin B 13 (see Scheme 3) – from the 5,6,5-indolines 2a or 2b. This required installation of the requisite butenyl sidechain, and conversion of the protected indoline to the free indole. For the former of these tasks, we recognised the synthetic value of the silane present in 2a/b, which enables a variety of sidechain attachment strategies. We first addressed Hiyama cross-coupling of 2b, which offers a direct route to the butenyl substituent and is an attractive alternative to other coupling methods (e.g. Stille, Suzuki) due to the low toxicity of silicon and its stability to multistep synthesis.9 To our knowledge, no Hiyama couplings between arylbenzyl dimethylsilanes and alkenyl halides have been reported, with only the reverse process being described (i.e. the coupling of alkenylbenzyldimethylsilanes with aryl halides).10
Standard conditions for the coupling of alkenyl benzyldimethylsilanes (TBAF, Pd2dba3·CHCl3 or Pd(dba)2),10 using either β-styrenyl iodide 7a or butenyl iodide 7b as the halide partner, afforded no cross-coupling product (Table 2, entries 1 and 2). As benzylsilanes are ‘safety-catch’ silanols, and indeed are hydrolysed to the latter on treatment with TBAF, alternative conditions for the coupling of alkenylsilanols11 were also investigated, without success (entry 3). In all of these trials, mixtures of silanol, disiloxane, and desilylated arene were recovered,12 suggesting that the aryl silanol revealed on unmasking of the benzylsilane was resistant to transmetallation. The addition of Ag2O has been reported by Hiyama to accelerate transmetallation,13 and we were delighted to find that the coupling of styrenyl iodide 7a under these conditions smoothly afforded the styrenyl trikentrin framework 8a (68%). Disappointingly, only desilylated arene was returned on attempted coupling with butenyl iodide7b, which for this study presented an insurmountable limitation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Alkenyl iodide | [Pd] cat. (mol%) | TBAF (equiv.) | Temp (°C) | Yielda (%) |
| a Isolated yield. b A mixture of silanol, disiloxane, and desilylated arene was recovered. c 1.1 equiv. Ag2O. d Desilylated 2b was isolated (67%). | |||||
| 1 | 7a | Pd2dba3·CHCl3 (2.5) or Pd(dba)2 (5) | 2.2 | 20 → 50 | —b |
| 2 | 7b | Pd2dba3·CHCl3 (2.5) | 2.2 | 20 → 50 | —b |
| 3 | 7a | (AllylPdCl)2 (2.5) | 2.2 | 20 → 50 | —b |
| 4 | 7a | Pd(PPh3)4 (5), Ag2Oc | 1.1 | 20 | 68 |
| 5 | 7b | Pd(PPh3)4 (5), Ag2Oc | 1.1 | 20 | —d |

 | ||
| Scheme 3 Synthesis of bis-desmethyl-trikentrin B. | ||
A more classical route to install the butenyl sidechain was thus developed (Scheme 3).14 Aryltrimethylsilane 2a was subjected to a Friedel–Crafts acylation, which proceeded with exclusive ipso-selectivity to give ketone9 (79%). This ketone then underwent a high-yielding reduction–dehydration sequence to deliver the targeted butenyl sidechain (8b). Completion of the synthesis now required indoline detosylation and oxidation to reveal the indole moiety. However, all attempts to oxidise 8b to the corresponding sulfonyl indole were unsuccessful, leading mainly to degradation.15 Inverting this sequence of events resolved this issue; although Mg/MeOH/sonication (which is usually effective for such detosylations)2a,16 effected partial deprotection (<25%), treatment of 8b with sodium naphthalenide gave the deprotected indoline 11 with high efficiency. Somewhat surprisingly, 11 underwent rapid aerobic decomposition, presumably due to the indoline-enhanced reactivity of the electron-rich styrene,17 and isolation of the pure indoline proved difficult. However, we were pleased to find that direct dehydrogenation of the crude indoline using Pd/C in degassed toluene completed the synthesis, giving bis-desmethyl-trikentrin 12 in good yield over the two steps.
In conclusion, we have developed a facile method for the preparation of azatricycles from bromoalkenyl ynamides. The reaction enables formation of five- to seven-membered rings, and offers an attractive alternative to cyclotrimerization strategies. The utility of this chemistry is demonstrated by installation of the trikentrin B alkenyl sidechain in a further four steps using Friedel–Crafts ipso-substitution of the arylsilane cyclization products. As an alternative, we report the first example of an alkenyl iodide/arylbenzylsilane Hiyama cross-coupling, which affords a styrenyl-trikentrin analogue.
We thank the EPSRC (EP/H025839/1, CDC; EP/E055273/1, Advanced Research Fellowship to E.A.A.), and Syngenta Ltd. for a studentship (to R.L.G.).
Notes and references
- For recent approaches to the trikentrins and herbindoles, see: (a) I. R. M. Tébéka, G. B. Longato, M. V. Craveiro, J. E. de Carvalho, A. L. T. G. Ruiz and L. F. Silva, Chem.–Eur. J., 2012, 18, 16890 CrossRef PubMed; (b) W. Liu, H. J. Lim and T. V. RajanBabu, J. Am. Chem. Soc., 2012, 134, 5496 CrossRef CAS PubMed; (c) N. Saito, T. Ichimaru and Y. Sato, Org. Lett., 2012, 14, 1914 CrossRef CAS PubMed . For a review and references to earlier syntheses: ; (d) L. F. Silva, Jr., M. V. Craveiro and I. R. M. Tébéka, Tetrahedron, 2010, 66, 3875 CrossRef PubMed.
- (a) R. L. Greenaway, C. D. Campbell, H. A. Chapman and E. A. Anderson, Adv. Synth. Catal., 2012, 354, 3187 CrossRef CAS; (b) R. L. Greenaway, C. D. Campbell, O. T. Holton, C. A. Russell and E. A. Anderson, Chem.–Eur. J., 2011, 17, 14366 CrossRef CAS PubMed; (c) P. R. Walker, C. D. Campbell, A. Suleman, G. Carr and E. A. Anderson, Angew. Chem., Int. Ed. DOI:10.1002/anie.201304186 . For other examples of ynamide carbopalladation, see: ; (d) S. Couty, B. Liegault, C. Meyer and J. Cossy, Tetrahedron, 2006, 62, 3882 CrossRef CAS PubMed; (e) S. Couty, C. Meyer and J. Cossy, Tetrahedron Lett., 2006, 47, 767 CrossRef CAS PubMed; (f) S. Couty, B. Liegault, C. Meyer and J. Cossy, Org. Lett., 2004, 6, 2511 CrossRef CAS PubMed.
- (a) M.-C. A. Cordonnier, S. B. J. Kan and E. A. Anderson, Chem. Commun., 2008, 5818 RSC; (b) S. S. Goh, H. Baars, B. Gockel and E. A. Anderson, Org. Lett., 2012, 14, 6278 CrossRef CAS PubMed . For recent reviews of palladium-catalyzed cascade processes, see: ; (c) T. Vlaar, E. Ruijter and R. V. A. Orru, Adv. Synth. Catal., 2011, 353, 809 CrossRef CAS; (d) E. A. Anderson, Org. Biomol. Chem., 2011, 9, 3997 RSC.
- For the development of bromoenediyne cyclizations, see: (a) W. M. Tokan, F. E. Meyer, S. Schweizer, P. J. Parsons and A. de Meijere, Eur. J. Org. Chem., 2008, 6152 CrossRef CAS; (b) H. Henniges, F. E. Meyer, U. Schick, F. Funke, P. J. Parsons and A. de Meijere, Tetrahedron, 1996, 52, 11545 CrossRef CAS; (c) E. Negishi, L. S. Harring, Z. Owczarczyk, M. M. Mohamud and M. Ay, Tetrahedron Lett., 1992, 33, 3253 CrossRef CAS.
- (a) B. Witulski and T. Stengel, Angew. Chem., Int. Ed., 1999, 38, 2426 CrossRef CAS; (b) B. Witulski, T. Stengel and J. M. Fernández-Hernández, Chem. Commun., 2000, 1965 RSC; (c) B. Witulski and C. Alayrac, Angew. Chem., Int. Ed., 2002, 41, 3281 CrossRef CAS . For recent examples of ynamides in [2+2+2] cyclization, see: ; (d) P. Garcia, Y. Evanno, P. George, M. Sevrin, G. Ricci, M. Malacria, C. Aubert and V. Gandon, Chem.–Eur. J., 2012, 18, 4337 CrossRef CAS PubMed; (e) F. Nissen, V. Richard, C. Alayrac and B. Witulski, Chem. Commun., 2011, 6656 RSC . See also ref. 1c.
- For a recent review, see: S. Kotha, E. Brahmachary and K. Lahiri, Eur. J. Org. Chem., 2005, 4741 CrossRef CAS . See also ref. 3b.
- For comprehensive reviews of ynamide chemistry, see: (a) Y. Zhang, K. A. DeKorver, H. Y. Li, A. G. Lohse, R. Hayashi, Z. Lu and R. P. Hsung, Chem. Rev., 2010, 110, 5064 CrossRef PubMed; (b) G. Evano, A. Coste and K. Jouvin, Angew. Chem., Int. Ed., 2010, 122, 2902 CrossRef.
- X. Zhang, Y. Zhang, J. Huang, R. P. Hsung, K. C. M. Kurtz, J. Oppenheimer, M. E. Petersen, I. K. Sagamanova, L. Shen and M. R. Tracey, J. Org. Chem., 2006, 71, 4170 CrossRef CAS PubMed.
- For reviews of Hiyama cross-coupling, see: (a) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845 RSC; (b) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC; (c) S. E. Denmark and J. H.-C. Liu, Angew. Chem., Int. Ed., 2010, 49, 2978 CrossRef CAS PubMed.
- (a) B. M. Trost, M. R. Machacek and Z. T. Ball, Org. Lett., 2003, 5, 1895 CrossRef CAS PubMed; (b) S. E. Denmark and S. A. Tymonko, J. Am. Chem. Soc., 2005, 127, 8004 CrossRef CAS PubMed; (c) S. E. Denmark and S. Fujimori, J. Am. Chem. Soc., 2005, 127, 8971 CrossRef CAS PubMed; (d) S. E. Denmark and J. H.-C. Liu, J. Am. Chem. Soc., 2007, 129, 3737 CrossRef CAS PubMed; (e) Y. Nishihara, D. Saito, K. Tanemura, S. Noyori and K. Takagi, Org. Lett., 2009, 11, 3546 CrossRef CAS PubMed; (f) C. Morrill and N. S. Mani, Org. Lett., 2007, 9, 1505 CrossRef CAS PubMed.
- (a) S. E. Denmark and L. Neuville, Org. Lett., 2000, 2, 3221 CrossRef CAS PubMed; (b) S. E. Denmark and W. J. Pan, J. Organomet. Chem., 2002, 653, 98 CrossRef CAS.
- See the ESI† for details.
- (a) K. Hirabayashi, J. Kawashima, Y. Nishihara, A. Mori and T. Hiyama, Org. Lett., 1999, 1, 299 CrossRef CAS; (b) K. Hirabayashi, A. Mori, J. Kawashima, M. Suguro, Y. Nishihara and T. Hiyama, J. Org. Chem., 2000, 65, 5342 CrossRef CAS.
- Alternative strategies, such as the use of ynamides already featuring the trikentrin B sidechain, were not investigated in this study, which targeted a system enabling skeletal diversification at a late stage.
- Oxidation using MnO2, Mn(OAc)3, DDQ, or AIBN/NBS led to complete degradation of material; Co(salen)/O2 led to no reaction.
- G. H. Lee, I. K. Youn, E. B. Choi, H. K. Lee, G. H. Yon, H. C. Yang and C. S. Pak, Curr. Org. Chem., 2004, 8, 1263 CrossRef CAS.
- Aldehyde-containing byproducts, presumably arising from cleavage of the alkene sidechain, were noted in this decomposition process.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and copies of 1H and 13C NMR spectra for novel compounds. See DOI: 10.1039/c3cc45634j |
| This journal is © The Royal Society of Chemistry 2014 |