
Microscale patterning of hydrogel stiffness through light-triggered uncaging of thiols†
Katarzyna A.
Mosiewicz‡
a,
Laura
Kolb‡
a,
André J.
van der Vlies‡
ab and
Matthias P.
Lutolf
*a
aInstitute of Bioengineering, School of Life Sciences and School of Engineering, Ecole Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland. E-mail: matthias.lutolf@epfl.ch
bDivision of Applied Chemistry, Graduate School of Engineering, Osaka University, Osaka 565-0871, Japan
First published on 4th September 2014
Abstract
Mammalian cell behavior is strongly influenced by physical and chemical cues originating from the extracellular matrix (ECM). In vivo, ECM signals are displayed in a spatiotemporally complex fashion, often composed as gradients and in concentration profiles that change in time. Most in vitro models to study the role of ECM signals in regulating cell behavior are limited in capturing this microenvironmental complexity, as they are static and homogeneous. In order to achieve a dynamic control of the physical properties of a hydrogel network, we here designed a chemical scheme to control poly(ethylene glycol) (PEG) hydrogel stiffness in space, time and intensity. Specifically, we combined caging chemistry and Michael-type addition to enable the light-triggered local control of hydrogel crosslinking density. Thiol moieties of one of the reactive PEG macromers undergoing crosslinking were equipped with caging groups to prevent their susceptibility to the counter-reactive vinyl sulfone groups on the termini of the complementary PEG macromers. Thus, the crosslinking density of the hydrogel network could be tuned by uncaging with light which directly translated into differential patterns of hydrogel stiffness. Using this approach, user-defined stiffness patterns in a range of soft tissue microenvironments (i.e. between 3–8 kPa) were obtained and shown to influence the migratory behavior of primary human mesenchymal stem cells (hMSC). Stiffness gradients in the higher range (5.5–8 kPa) were able to elicit durotaxis towards the more densely crosslinked regions, whereas those in the lower range (3–5.5 kPa) showed no significant directional preference in hMSC migration. Our patterning tool should be useful for the manipulation of cell fate in various other contexts.
Introduction
In the last two decades, synthetic hydrogels have emerged as versatile biomaterials for tissue engineering and cell biology.1–5 Their high water content and tunable physicochemical properties have allowed researchers to mimic the physical properties of specific ECMs of native tissues to study cell behaviour in vitro.6,7 However, most hydrogels are static and lack the possibility to recapitulate the dynamic properties of natural ECMs that are composed of multiple graded signalling cues and concentration profiles that are also changing in time. Indeed, this spatiotemporally tightly controlled display of cell-instructive signals is, for example, crucial in directing cell behaviour during development.7–9In order to mimic dynamic ECM properties, new families of light-responsive synthetic hydrogels have been developed.10–12 A major focus has been on modulating by a light beam the stiffness of such polymer networks at the micrometer scale and at a desired time during an experiment.13 Indeed, just like in the case of biochemical signals, microscale heterogeneities in ECM stiffness have been shown to be involved in regulating cell fate during embryonic development, for example.14–16
One widely used technique to generate such gels is the localized UV-induced radical crosslinking of methacrylate-, acrylate- or norbornene functionalized polymers.17–20 The application of this scheme has for example been used to locally alter crosslinking densities of hydrogels such as to generate stiffness gradients and manipulate cell behaviour in 2D and 3D.21–24 However, a drawback of radical reactions is their relatively poorly controllable crosslinking kinetics, which can lead to sharp rather than smooth gradients. Furthermore, the generated free radicals can compromise cell viability in 3D contexts. Although recent advances such as improved UV photoinitiators (e.g. Irgacure 2959, I2959, and lithium phenyl-2,4,6-trimethylbenzoylphosphinate, LAP) and the new rapid step-growth mechanism of thiol-norbornene show low cytotoxicity and little effect on protein bioactivity, the need for radical-free photoinduction still persists, especially for radical-sensitive cell types.25–27 To overcome these limitations, researchers have begun to employ photo-releasing or ‘caging’ chemistries in order to spatiotemporally modify hydrogel properties.28,29 By light-induced cleavage of a bulky protecting group from a reactive site, caging technology allows to modulate biomolecular interactions and is now routinely used to investigate molecular processes in biology such as protein trafficking, and has been employed to locally control biochemical properties of hydrogels.29–31,32–34 Anseth et al. have for example adapted this concept for hydrogel engineering by local light-mediated cleavage of a 2-nitrobenzyl linker in the PEG backbone to decrease hydrogel stiffness upon UV (365 nm), visible (405 nm) or two-photon (740 nm) light exposure.28 This photo-degradation scheme was successfully applied to control the spreading of hMSCs within light-eroded channels. Although photo-degradation is an excellent approach for spatiotemporal hydrogel manipulation, it allows for altering gel features only in a subtractive rather than in an additive manner. Thus matrix stiffening is not possible using this approach. Considering this, we here introduce a novel scheme to control the intensity and location of stiffness in synthetic hydrogels. Our approach is based on photo-labile caging groups to temporally mask one of the functional groups involved in hydrogel crosslinking (Fig. 1). Hence, local light exposure by uncaging causes reactivation of the functional group that can then undergo crosslinking, creating local patterns of higher stiffness. We reasoned that an advantage of this approach would lie in its simplicity, specificity and the precision by which patterns of any desired shape and intensity variation could be generated.
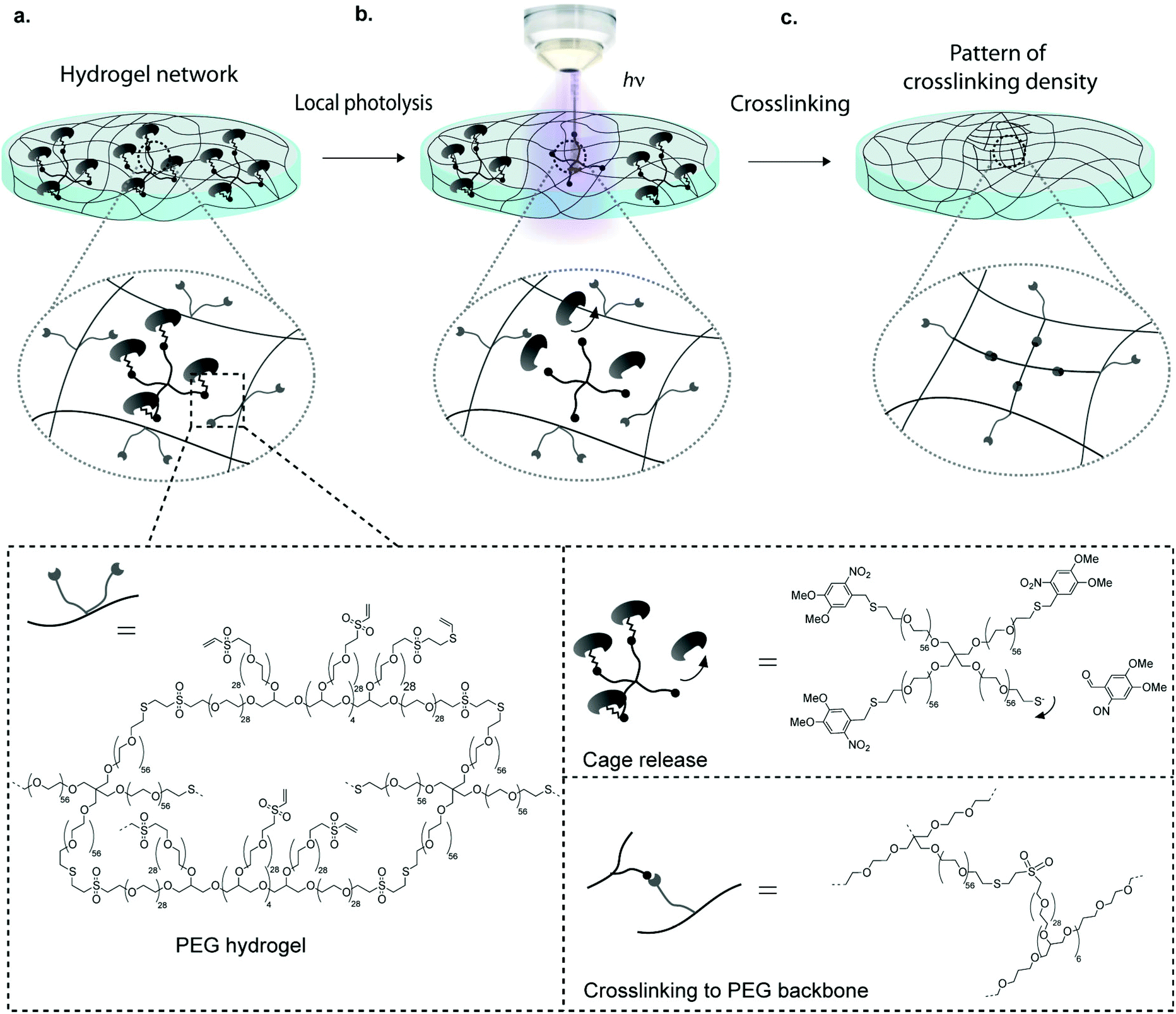 | ||
| Fig. 1 Concept of light-mediated Michael-type addition for spatiotemporally controlled hydrogel crosslinking; a, A thiol-containing macromer is protected with photo-labile caging group and is dissolved in a hydrogel network comprising free vinyl sulfone groups; b, localized laser illumination causes photocleavage of the caging groups reactivating thiols; c, activated thiol-containing PEGs react to vinyl sulfone via Michael-type addition reaction creating localized stiffness patterns. | ||
To implement this concept, we chose a chemical hydrogel crosslinking scheme that is based on the well-established Michael-type addition of thiols onto unsaturated vinyl sulfone groups.35–37 This reaction is fast and reasonably specific, that is, at physiological pH competing reactions of vinyl sulfones with other nucleophiles such as amines are considerably slower.5,38 We reasoned that by temporally masking thiols with a photo-labile caging group, it should be possible to obtain light-dependent hydrogel crosslinking (Fig. 1). Here we show that patterning gels by photo-controlled Michael-type addition can be translated to highly localized stiffness patterns of broadly tunable shape and stiffness range. Using this approach, user-defined stiffness gradients were obtained and shown to influence the migratory behavior of hMSCs. It should be noted that during the preparation of this manuscript a somewhat similar approach of photo-controlled Michael-type addition to control hydrogel size and shape was published.39 However, the authors used a different caging group as well as maleimide, a functional group that is prone to hydrolysis, in contrast to the much more stable vinyl sulfone group used here.
Experimental
Materials
Standard chemicals were purchased from Fluka AG or Sigma-Aldrich AG (Buchs, CH). Alexa Fluor 488 C5 maleimide was from Invitrogen. Hydroxyl-terminated 8arm-PEG (Mw = 10 kDa), thiol-functionalized 4arm-PEG-SH (Mw = 20 kDa) and linear methoxy-PEG-SH (Mw = 5 kDa) was purchased from NOF Corporation (Japan). PEG vinyl sulfone (PEG-VS) was produced and characterized as described elsewhere.40 NMR spectra were recorded on a Bruker 400 MHz spectrometer. Reverse phase HPLC was performed on a Waters 2790 separation module with a binary gradient of water/TFA and acetonitrile coupled to UV detector (Photodiode Array Detector, Waters Corp. model 2996, Baden-Dättwil, Switzerland). Cell adhesion peptide: Ac-GRCGRDSPG-NH2 was custom-made by GL Biochem (Shanghai).The AFM to measure elastic moduli was Nanowizard II (JPK Instruments, Germany) in Force Mode, mounted on an epifluorescence microscope (Axiovert 100, Zeiss). We used a spherical silicon AFM cantilever (radius of curvature: 1 μm, nominal spring constants: 0.02 N m−1, Microlevers, Nanoworld, Medium Sphere Type CONT). Elastic moduli were calculated using the JPK Data Processing software (JPK Instruments, Germany).
The uncaging efficiencies for all the caged PEG compounds was determined based on area of peaks corresponding to caged PEG illuminated for indicated time with UV light. The data of photodeprotection was fitted to the first-order rate equation42:
| [c]0 − [c]t = [c]0 (1 − e−kt) |
| t1/2 = ln(2)/k |
Results and discussion
Selection of caging chemistry for rapid photo-release in the visible light absorption spectrum
We chose three thiol-caging groups based on the key criteria of rapid photo-release kinetics, in order to obtain patterning without extensive illumination of the hydrogel sample, as well as cell-friendly, near UV-VIS wavelengths such as to be able to apply the system in the presence of cells with good depth penetration. Consequently, caging groups with promising characteristics for hydrogel patterning applications were chosen based on the criteria ϕε > 100 (with ε = extinction coefficient, corresponding to the strength of light absorption at a given wavelength; ϕ = quantum yield, corresponding to the efficiency of a caused effect by absorbed light).46,47The cages o-nitrobenzyl (NB) and its derivative dimethoxy nitrobenzyl (DMNB) (Fig. 2a) belong to the o-nitrobenzyl family and are the most commonly used photo-labile molecules for controlling biochemical processes. Their main advantages over other cages are the fast deprotection rate and the sensitivity to near UV-VIS. Furthermore, NB is reported to possess higher deprotection rate but at lower wavelengths, while DMNB was reported to have excellent absorption in the range of near UV-VIS light.31,47–49
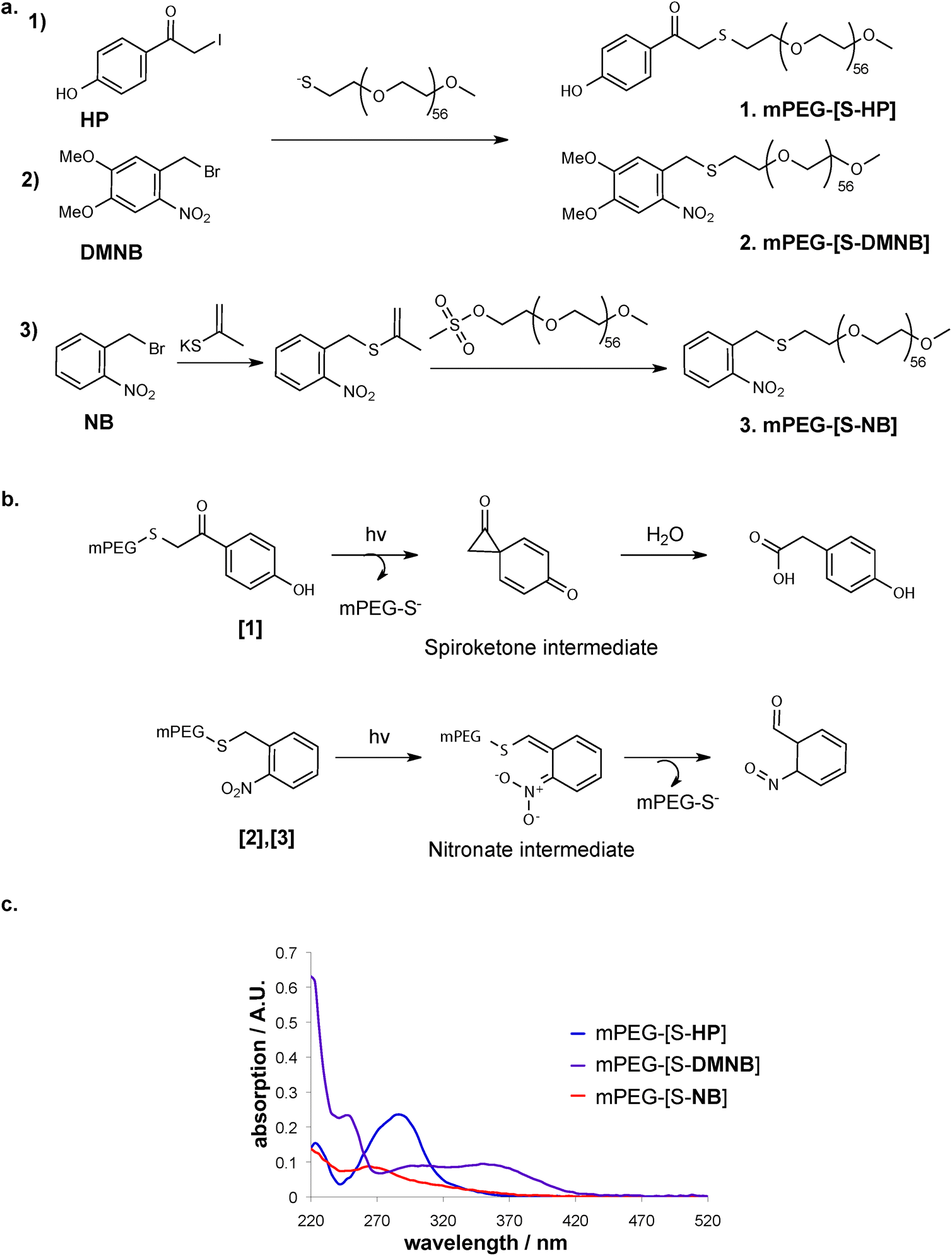 | ||
| Fig. 2 Soluble models of caged linear methoxy-PEG-[S-]; a, synthetic schemes involving the caging groups (1): 1-(4-hydroxyphenyl)-2-iodoethanone (HP), (2): 1-(bromomethyl)-2-nitrobenzene (NB), (3): 1-(bromomethyl)-4,5-dimethoxy-2-nitrobenzene (DMNB); b, simplified mechanism of photolysis reaction for hydroxyphenacyl group (upper reaction) and nitrobenzene family of cages (lower panel); c, measured UV absorption spectra of the cages. | ||
The third caging group that was selected belongs to the family of p-hydroxyphenacyl (HP) cages. Specifically, we used hydroxyphenacetyl iodide (Fig. 2a) that is suitable for conjugation to PEG due to iodide as a very good leaving group.50 HP cages have an advantageous photolytic chemical mechanism compared to other cage, in which the photoactivation happens immediately in the very first step of photolysis (Fig. 2b).
We first investigated the rate of photo-release of these cages when attached to a mono-functional methoxy-PEG-thiol (mPEG-SH, 5 kDa) as a soluble model system for the crosslinked PEG hydrogel. The different cages were conjugated to the sulfhydryl group of the mPEG-SH (Fig. 2a) leading to caged mPEG-SH (mPEG-[S-]). When the UV absorption of the obtained caged linear PEGs was compared spectrophotometrically (Fig. 2c), a broad absorption spectrum of DMNB reaching visible light was observed, whereas in the case of HP high absorption was only detected around 300 nm.
To analyze the kinetics of light-induced cleavage of the cages, we utilized an HPLC-based method that allowed quantification of the rate of cage release. Illumination was performed using a UV mercury lamp for different time periods and the samples were injected into an analytical C18 column (Fig. 3a). This analysis revealed the light-dependent decrease in peaks corresponding to caged PEG (Fig. 3b). By measuring the decrease in signal intensity from caged mPEG-[S-] and fitting a linear regression, we obtained the half-lives of photolysis (Fig. 3c, ESI† Table 1). The slowest reaction was obtained for mPEG-[S-HP] with t1/2 of 33.2 seconds. Much better performance was obtained for the nitrobenzyl cages, ranging from t1/2 = 4.7 seconds for NB to t1/2 = 11.9 seconds for DMNB. Based on these results we chose DMNB for further studies.
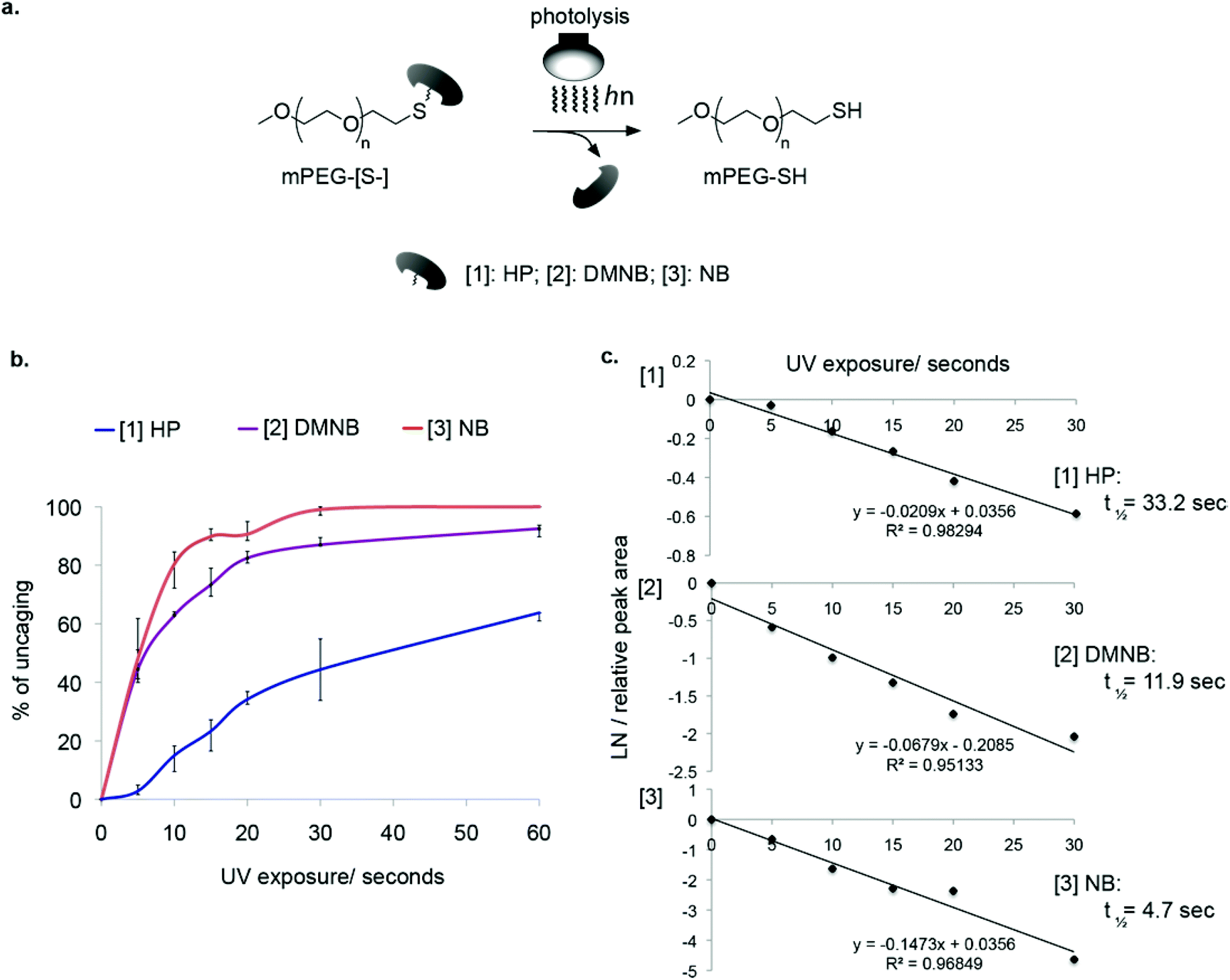 | ||
Fig. 3 Model for studying photolysis kinetics of the caged-PEGs; a, Schematic of photolysis leading to reactive thiols; b, Uncaging kinetics determined by RP-HPLC, revealing the rate of photo-release of the cages (– HP, [2] – NB, [3]– DMNB); c, Analysis of photo-release kinetics fitted to first-order rate equation. The slope of the linear regression gives a value for −k and, consequently, half-life of photolysis was obtained (t1/2 = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2/k) corresponding to HP: t1/2 = 33.2, NB: t1/2 = 4.7, DMNB: t1/2 = 11.9 seconds. 2/k) corresponding to HP: t1/2 = 33.2, NB: t1/2 = 4.7, DMNB: t1/2 = 11.9 seconds. | ||
Photo-controllable Michael-type addition affords precise spatial control of hydrogel stiffness
To render hydrogel crosslinking via Michael-type addition susceptible to light, 4arm-PEG-SH macromers (10 kDa) were modified with DMNB. Mixing of two aqueous solutions containing caged 4arm-PEG-[S-]46 and 8arm-PEG-VS at equal stoichiometry of functional groups did not result in any gelation. In contrast, illumination of these precursors with a confocal microscope laser at defined regions of interests (ROI) resulted in microscale hydrogel patches (Fig. 4). This experiment proves that thiol uncaging is very efficient and a combination of photochemistry and Michael-type addition affords precise microscale synthesis of hydrogels. Furthermore, the use of confocal lasers allows for scanning with high resolution practically any designed patterns by simply drawing user-defined shapes. The current setting allows a patterning down to single cell resolution of about 10 μm (data not shown). In theory, the actual limit to the resolution is the objective or the microscope that is used, respectively, and thus can be further increased according to the needs of the user.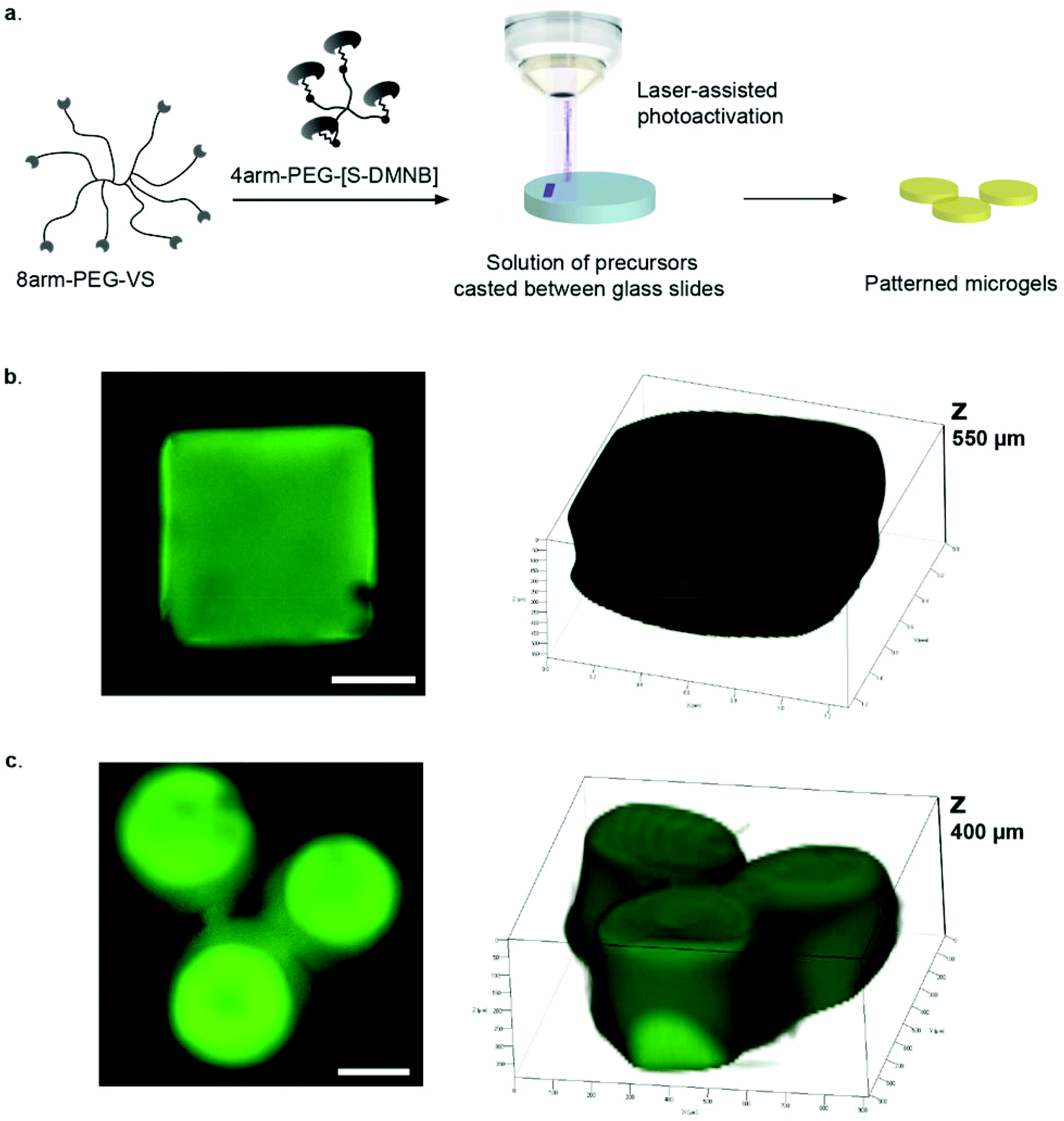 | ||
| Fig. 4 a, Concept of photo-controllable Michael-type addition for bulk hydrogel crosslinking. An aqueous solution of PEG-vinylsulfone (VS) and caged PEG-[S-DMNB] is non-reactive due to caged thiols; b,c, Fluorescent microgels obtained by spots of LSM confocal laser; b, Cubic hydrogel (scale bar = 300 μm); c, Smaller cylindrical hydrogels (scale bar = 200 μm). | ||
We next focused on locally modulating stiffness in already formed hydrogel samples according to the scheme indicated in Fig. 1. To this end, soluble caged 4arm-PEG-[S-] was added to a pre-made hydrogel having 50% excess vinyl sulfone groups available for crosslinking with equimolar amounts of photo-activated thiols (Fig. 5a). The pre-made hydrogels were then exposed to a confocal laser (405 nm diode laser, 30 mW) using defined laser intensities. Specifically, 100% of the maximal laser power for 840 μm × 840 μm square and a series of different intensities corresponding to 12.5, 25, 50 and 100% of the maximal laser power assigned to rectangular ROIs of 420 μm width were used. The time of laser irradiation was equal to the sum of 50 iterations of laser scanning. Notably, patterns were readily visible by phase contrast microscopy (Fig. 5b), whereas in a negative control lacking 4arm-PEG-[S-] no pattern was formed. Moreover, it was possible to visualize patterned areas by fluorescently labeling thiols using a maleimide dye and confocal imaging (Fig. 5c). Importantly, the intensity of the fluorescent signal obtained by maleimide-based labeling directly correlated with the laser intensity. Again, no fluorescent pattern was observed in the case of a negative control missing caged 4arm-PEG-[S-]. These data confirm the successful photolysis of caging groups from 4arm-PEG-[S-] macromers in an intensity-dependent manner.
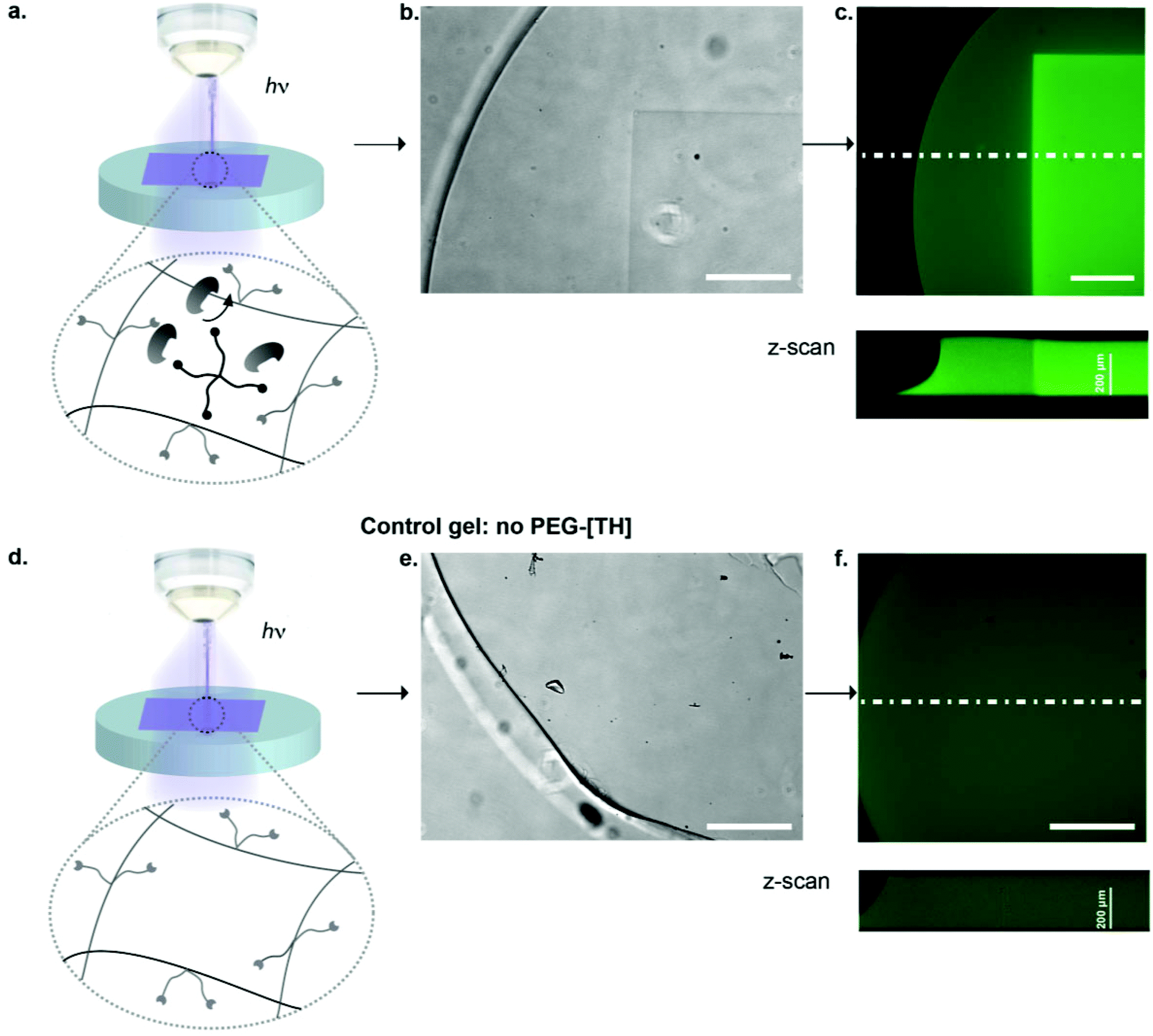 | ||
| Fig. 5 Generation of physical heterogeneities in hydrogel; a, Schemes of photo-patterning hydrogel crosslink densities to locally stiffen hydrogels; b, Phase contrast image of hydrogel pattern obtained after photo-activated crosslinking; c, Pattern visualization by fluorescent labeling with thiol-reactive, maleimide-functionalized dye; d, Negative control of photo-patterning procedure with Michael-type addition hydrogel that lacks the photo-labile PEG-[S-DMNB] linker during photolysis; e–f, No patterns can be observed. Scale bars: 200 μm in b–e, 300 μm in c–f. | ||
Next, we measured the elasticity of the obtained micropatterns on the surface of PEG hydrogels using atomic force microscopy (AFM).43 We analyzed each indentation force-response curve with a point-wise method, such that the depth-dependent, apparent elastic modulus could be computed as a function of indentation depth using a modified Hertz model.51 These measurements showed a discrete microstiffness gradient, ranging from 3.3 (±0.1) to 8.2 (±0.3) kPa measured in micro-squares of 420 μm × 420 μm (Fig. 6). We observed minimal stiffness variation within each section of the discrete gradient. Therefore, patterning gels by photo-controlled Michael-type-addition is not limited to the production of simple binary or linear changes in stiffness, as stiffness patterns of practically any desired shape and stiffness range can be used.
 | ||
| Fig. 6 AFM to quantify local hydrogel stiffness. a, Design of regions of interest (ROIs) for laser exposure; b, Hydrogel patterns in hydrogel visualized fluorescently by labeling with a fluorescent dye (scale bar = 200 μm); c, Obtained mean values of elastic moduli measured by AFM. | ||
Directed hMSC migration on photomodulated stiffness gradients
To demonstrate the utility of our system for biological applications, we tested the hydrogel stiffness-patterning platform in a cellular context. We examined the influence of local gel crosslinking density on the migratory behavior of hMSCs. It has been well established that these cells can respond to substrate elasticity by changing morphology, motility and even lineage specification.21,22,52 Lo and others have already demonstrated that many different cell types undergo durotaxis, that is, the directed migration along a stiffness gradient.22,53–56 Specifically, guiding hMSC migration by mimicking these heterogeneous elasticity features of the substrate by complex and dynamic hydrogel designs might be a valuable tool to study mechanisms of wound healing, morphogenesis and cancer progression, and eventually exploit them in tissue engineering devices.To probe the migratory behavior of hMSCs on a discrete stiffness gradient, we designed regions of interest (ROIs) with 20 different stiffness ranges (Fig. 7a), with elastic moduli in the range of soft tissues7 (between 3 and 8 kPa) and spanning a region of approximately 1000 × 900 μm. In addition, the PEG hydrogels were rendered cell-adhesive by tethering to the gels 1 mM of the fibronectin-derived adhesion peptide RGDSP during initial crosslinking to ensure homogenous distribution. When hMSCs were seeded at a cell density of 1000 cells cm−2, they initially attached homogenously on the hydrogel surface (Fig. 7b). Single cell migration tracks on the patterns were imaged by time-lapse microscopy and analyzed by the TrackMate plugin (Fiji) (Fig. 7c and ESI Movie†). The mean velocity was found to be significantly increased on the softer area of the gradient (ESI Fig. 1a†). Overall, hMSCs seeded on the mechanically modulated area of increasing substrate rigidity showed a loss of random motility, i.e. cell migration seemed to be more directionally biased in comparison to the softer non-patterned outside (ESI Fig. 1b†). Another means to assess directional migration is the persistence time which was not significantly different in our setup (data not shown).57,58 Notably, the assessment of the direction of the displacement by the analysis of the orientation angle of the net displacement revealed that the stiffness gradient of 5.5 kPa onwards was able to elicit durotaxis towards the more densely crosslinked regions (Fig. 7d and e). In contrast, cell migration on the stiffness gradient in the lower range (3–5.5 kPa) showed no significant directional preference. This finding indicated a threshold of about 5.5 kPa to initiate durotactic behavior in hMSCs in our system, i.e. in a modulus range-dependent manner with constant gradient strength. These initial data reveal a significant effect of local, gradual change of the crosslinking density on cell migration. Stiffness levels and steepness of a gradient are important regulators in durotaxis of cells and our platform should provide means for the further investigation of these parameters.22,55,56
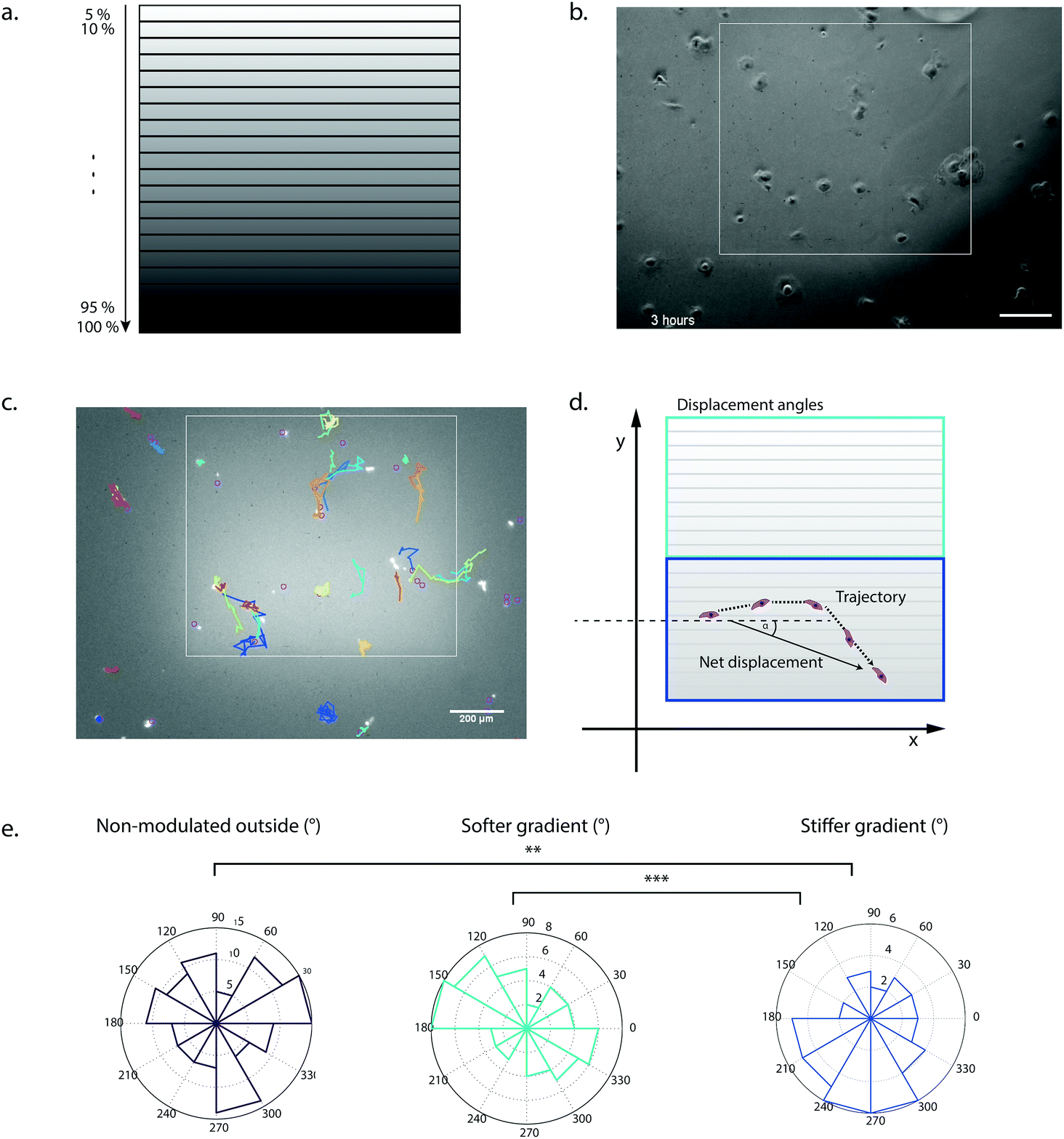 | ||
| Fig. 7 hMSC behavior on stiffness patterns. a, Design of patterns by translation of stepwise increasing laser intensity into regions of increasing stiffness. b, hMSCs are cultured on top of photopatterned hydrogels and imaged by time-lapse microscopy for 36 hours. The gradient was visualized by a fluorescent dye and borders are indicated by a white frame. hMSCs initially attached homogenously on the hydrogel surface (Scale bar = 200 μm). c, Single cell nuclei are stained by Celllight and are tracked by the TrackMate plugin (Fiji). Single cell tracks are indicated by different colors. d, Cell tracks for the non-modulated soft area, on patterned soft areas (3–5.5 kPa, light blue) and stiffer areas (5.5–8 kPa, dark blue) are analyzed. Displacement orientation angle α is defined by the angle between the direction of the net displacement of a cell and the long side of the designed ROIs of the gradient. e, Displacement orientation angles are significantly different between the stiffer area and the non-modulated and patterned softer area respectively and mainly oriented towards the stiffer region indicating that the cells undergo durotaxis from 5.5 kPa onwards (**p < 0.01, ***p < 0.001, error bars represent s.d., n = 3 independent experiments, 113, 51 and 42 cells were tracked for the outside, softer gradient and stiffer gradient regions respectively). | ||
Taken together, we think that the combination of caging chemistry and Michael-type addition is an efficient strategy for the light-triggered control of hydrogel stiffness. This patterning concept should be also useful in the context of 3D cell cultures to manipulate the behaviour of various cell types.
Acknowledgements
We thank Lara Buscemi and Simone Allazetta for help with AFM, Vincent Pickenhahn for help with the synthesis of the caged products, Olivier Burri for developing a Matlab script for analysis of the TrackMate data, and Yoji Tabata for the displacement orientation angle analysis. This work was supported by the Swiss National Science Foundation grants CR32I3_125426 and CR23I2_125290, an ERC grant (#311422), the EU FP7 Large-scale integrating project ‘BIODESIGN’ (FP7-NMP-2010-LARGE-4).Notes and references
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Designing materials to direct stem-cell fate, Nature, 2009, 462, 433–441, DOI:10.1038/nature08602.
- J. A. Burdick and G. Vunjak-Novakovic, Engineered Microenvironments for Controlled Stem Cell Differentiation, Tissue Eng., Part A, 2009, 15, 205–219, DOI:10.1089/Ten.Tea.2008.0131.
- R. Langer and D. A. Tirrell, Designing materials for biology and medicine, Nature, 2004, 428, 487–492, DOI:10.1038/Nature02388.
- J. P. Vacanti and R. Langer, Tissue engineering: the design and fabrication of living replacement devices for surgical reconstruction and transplantation, Lancet, 1999, 354, SI32–SI34 CrossRef.
- S. Q. Liu, et al. Synthetic hydrogels for controlled stem cell differentiation, Soft Matter, 2010, 6, 67–81, 10.1039/B916705f.
- M. P. Lutolf, et al. Repair of bone defects using synthetic mimetics of collagenous extracellular matrices, Nat. Biotechnol., 2003, 21, 513–518 CrossRef CAS PubMed.
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Growth Factors, Matrices, and Forces Combine and Control Stem Cells, Science, 2009, 324, 1673–1677, DOI:10.1126/Science.1171643.
- J. B. Gurdon and P. Y. Bourillot, Morphogen gradient interpretation, Nature, 2001, 413, 797–803 CrossRef CAS PubMed.
- F. Chowdhury, et al. Material properties of the cell dictate stress-induced spreading and differentiation in embryonic stem cells, Nat. Mat., 2010, 9, 82–88, DOI:10.1038/Nmat2563.
- J. S. Katz and J. A. Burdick, Light-Responsive Biomaterials: Development and Applications, Macromol. Biosci., 2010, 10, 339–348, DOI:10.1002/Mabi.200900297.
- M. P. Lutolf, Spotlight on hydrogels, Nat. Mater., 2009, 8, 451–453 CrossRef CAS PubMed.
- J. A. Burdick, Bioengineering Cellular Control in Two Clicks, Nature, 2009, 460, 469–470, DOI:10.1038/460469a.
- B. P. Chan, Biomedical Applications of Photochemistry, Tissue Eng., Part B, 2010, 16, 509–522, DOI:10.1089/Ten.Teb.2009.0797.
- K. K. Parker and D. E. Ingber, Extracellular matrix, mechanotransduction and structural hierarchies in heart tissue engineering, Philos. Trans. R. Soc. Lond. B. Biol. Sci., 2007, 362, 1267–1279, DOI:10.1098/Rstb.2007.2114.
- M. S. Steinberg, Differential adhesion in morphogenesis: a modern view, Curr. Opin. Genet. Dev., 2007, 17, 281–286, DOI:10.1016/J.Gde.2007.05.002.
- V. Terraciano, et al. Differential response of adult and embryonic mesenchymal progenitor cells to mechanical compression in hydrogels, Stem Cells, 2007, 25, 2730–2738, DOI:10.1634/Stemcells.2007-0228.
- S. Nemir, H. N. Hayenga and J. L. West, PEGDA Hydrogels With Patterned Elasticity: Novel Tools for the Study of Cell Response to Substrate Rigidity, Biotechnol. Bioeng., 2010, 105, 636–644 CrossRef CAS PubMed.
- M. T. Thompson, et al. Biochemical functionalization of polymeric cell substrata can alter mechanical compliance, Biomacromolecules, 2006, 7, 1990–1995, DOI:10.1021/Bm060146b.
- K. T. Nguyen and J. L. West, Photopolymerizable hydrogels for tissue engineering applications, Biomaterials, 2002, 23, 4307–4314 CrossRef CAS.
- B. D. Fairbanks, et al. A Versatile Synthetic Extracellular Matrix Mimic via Thiol-Norbornene Photopolymerization, Adv. Mater., 2009, 21, 5005–5010, DOI:10.1002/adma.200901808.
- R. A. Marklein and J. A. Burdick, Spatially controlled hydrogel mechanics to modulate stem cell interactions, Soft Matter, 2010, 6, 136–143, 10.1039/B916933d.
- J. R. Tse and A. J. Engler, Stiffness Gradients Mimicking In Vivo Tissue Variation Regulate Mesenchymal Stem Cell Fate, PLoS One, 2011, 6 DOI:10.1371/journal.pone.0015978.
- M. Guvendiren and J. A. Burdick, Stiffening hydrogels to probe short- and long-term cellular responses to dynamic mechanics, Nat. Commun., 2012, 3, 792, DOI:10.1038/ncomms1792.
- W. M. Gramlich, I. L. Kim and J. A. Burdick, Synthesis and orthogonal photopatterning of hyaluronic acid hydrogels with thiol-norbornene chemistry, Biomaterials, 2013, 34, 9803–9811, DOI:10.1016/j.biomaterials.2013.08.089.
- C. G. Williams, A. N. Malik, T. K. Kim, P. N. Manson and J. H. Elisseeff, Variable cytocompatibility of six cell lines with photoinitiators used for polymerizing hydrogels and cell encapsulation, Biomaterials, 2005, 26, 1211–1218, DOI:10.1016/j.biomaterials.2004.04.024.
- B. D. Fairbanks, M. P. Schwartz, C. N. Bowman and K. S. Anseth, Photoinitiated polymerization of PEG-diacrylate with lithium phenyl-2,4,6-trimethylbenzoylphosphinate: polymerization rate and cytocompatibility, Biomaterials, 2009, 30, 6702–6707, DOI:10.1016/j.biomaterials.2009.08.055.
- J. D. McCall and K. S. Anseth, Thiol-ene photopolymerizations provide a facile method to encapsulate proteins and maintain their bioactivity, Biomacromolecules, 2012, 13, 2410–2417, DOI:10.1021/bm300671s.
- A. Kloxin, A. M. Kasko, C. N. Salinas and K. Anseth, Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- Y. Luo and M. S. Shoichet, A photolabile hydrogel for guided three-dimensional cell growth and migration, Nat. Mater., 2004, 3, 249–253 CrossRef CAS PubMed.
- C. Campos, M. Kamiya, S. Banala, K. Johnsson and M. Gonzalez-Gaitan, Labelling Cell Structures and Tracking Cell Lineage in Zebrafish Using SNAP-Tag, Dev. Dyn., 2011, 240, 820–827, DOI:10.1002/Dvdy.22574.
- D. Maurel, S. Banala, T. Laroche and K. Johnsson, Photoactivatable and Photoconvertible Fluorescent Probes for Protein Labeling, ACS Chem. Biol., 2010, 5, 507–516, DOI:10.1021/Cb1000229.
- B. N. Goguen and B. Imperiali, Chemical Tools for Studying Directed Cell Migration, ACS Chem. Biol., 2011, 6, 1164–1174, DOI:10.1021/Cb200299k.
- R. G. Wylie, et al. Spatially controlled simultaneous patterning of multiple growth factors in three-dimensional hydrogels, Nat. Mater., 2011, 10, 799–806, DOI:10.1038/Nmat3101.
- K. A. Mosiewicz, et al. In situ cell manipulation through enzymatic hydrogel photopatterning, Nat. Mater., 2013, 12, 1072–1078, DOI:10.1038/nmat3766.
- M. P. Lutolf, N. Tirelli, S. Cerritelli, L. Cavalli and J. A. Hubbell, Systematic modulation of Michael-type reactivity of thiols through the use of charged amino acids, Bioconjugate Chem., 2001, 12, 1051–1056, DOI:10.1021/Bc015519e.
- K. Bott, et al. The effect of matrix characteristics on fibroblast proliferation in 3D gels, Biomaterials, 2010, 31, 8454–8464, DOI:10.1016/J.Biomaterials.2010.07.046.
- P. M. Gilbert, et al. Substrate Elasticity Regulates Skeletal Muscle Stem Cell Self-Renewal in Culture, Science, 2010, 329, 1078–1081, DOI:10.1126/Science.1191035.
- D. L. Elbert, A. B. Pratt, M. P. Lutolf, S. Halstenberg and J. A. Hubbell, Protein delivery from materials formed by self-selective conjugate addition reactions, J. Controlled Release, 2001, 76, 11–25 CrossRef CAS.
- Z. Liu, et al. Spatiotemporally Controllable and Cytocompatible Approach Builds 3D Cell Culture Matrix by Photo-Uncaged-Thiol Michael Addition Reaction, Adv. Mater., 2014, 26, 3912–3917, DOI:10.1002/adma.201306061.
- M. P. Lutolf and J. A. Hubbell, Synthesis and physicochemical characterization of end-linked poly(ethylene glycol)-co-peptide hydrogels formed by Michael-type addition, Biomacromolecules, 2003, 4, 713–722, DOI:10.1021/Bm025744e.
- M. L. N. Rao and D. N. Jadhav, Metal catalyst-free direct alpha-iodination of ketones with molecular iodine, Tetrahedron Lett., 2006, 47, 6883–6886, DOI:10.1016/J.Tetlet.2006.07.057.
- W. S. Dillmore, M. N. Yousaf and M. Mrksich, A photochemical method for patterning the immobilization of ligands and cells to self-assembled monolayers, Langmuir, 2004, 20, 7223–7231, DOI:10.1021/La049826v.
- Y. K. Cheung, et al. Microscale Control of Stiffness in a Cell-Adhesive Substrate Using Microfluidics-Based Lithography, Angew. Chem., Int. Ed, 2009, 48, 7188–7192, DOI:10.1002/Anie.200900807.
- O. V. Semenov, et al. Multipotent mesenchymal stem cells from human placenta: critical parameters for isolation and maintenance of stemness after isolation, Am. J. Obstet. Gynecol., 2010, 202, 193, DOI:10.1016/j.ajog.2009.10.869.
- R. Pankov, et al. A Rac switch regulates random versus directionally persistent cell migration, J. Cell Biol., 2005, 170, 793–802, DOI:10.1083/jcb.200503152.
- G. C. R. Ellis-Davies, Caged compounds: photorelease technology for control of cellular chemistry and physiology, Nat. Methods, 2007, 4, 619–628, DOI:10.1038/Nmeth1072.
- M. Goeldner and R. S. Givens, Dynamic Studies in Biology: Phototriggers, Photoswitches and Caged Biomolecules, Wiley-VCH, 2005 Search PubMed.
- J. W. Karpen, A. L. Zimmerman, L. Stryer and D. A. Baylor, Gating Kinetics of the Cyclic-Gmp-Activated Channel of Retinal Rods - Flash-Photolysis and Voltage-Jump Studies, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 1287–1291 CrossRef CAS.
- Y. R. Zhao, et al. New caged coumarin fluorophores with extraordinary uncaging cross sections suitable for biological imaging applications, J. Am. Chem. Soc., 2004, 126, 4653–4663, DOI:10.1021/Ja036958m.
- R. S. Givens, et al. New phototriggers 9: p-hydroxyphenacyl as a C-terminal photoremovable protecting group for oligopeptides, J. Am. Chem. Soc., 2000, 122, 2687–2697 CrossRef CAS.
- K. D. Costa, A. J. Sim and F. C. P. Yin, Non-Hertzian approach to analyzing mechanical properties of endothelial cells probed by atomic force microscopy, J. Biomech. Eng.–Trans. ASME, 2006, 128, 176–184, DOI:10.1115/1.2165690.
- S. Gobaa, et al. Artificial niche microarrays for probing single stem cell fate in high throughput, Nat. Methods, 2011, 8, 949–955, DOI:10.1038/Nmeth.1732.
- C. M. Lo, H. B. Wang, M. Dembo and Y. L. Wang, Cell movement is guided by the rigidity of the substrate, Biophys. J., 2000, 79, 144–152 CrossRef CAS.
- J. Y. Wong, A. Velasco, P. Rajagopalan and Q. Pham, Directed Movement of Vascular Smooth Muscle Cells on Gradient-Compliant Hydrogels, Langmuir, 2003, 19, 1908–1913, DOI:10.1021/la026403p.
- L. G. Vincent, Y. S. Choi, B. Alonso-Latorre, J. C. del Alamo and A. J. Engler, Mesenchymal stem cell durotaxis depends on substrate stiffness gradient strength, Biotechnol. J., 2013, 8, 472–484, DOI:10.1002/biot.201200205.
- B. C. Isenberg, P. A. Dimilla, M. Walker, S. Kim and J. Y. Wong, Vascular smooth muscle cell durotaxis depends on substrate stiffness gradient strength, Biophys. J., 2009, 97, 1313–1322, DOI:10.1016/j.bpj.2009.06.021.
- M. F. Ware, A. Wells and D. A. Lauffenburger, Epidermal growth factor alters fibroblast migration speed and directional persistence reciprocally and in a matrix-dependent manner, J. Cell Sci., 1998, 111(Pt 16), 2423–2432 CAS.
- C. L. Stokes, D. A. Lauffenburger and S. K. Williams, Migration of individual microvessel endothelial cells: stochastic model and parameter measurement, J. Cell Sci., 1991, 99(Pt 2), 419–430 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4bm00262h |
| ‡ These authors contributed equally to the work presented. |
| This journal is © The Royal Society of Chemistry 2014 |