
Combination of magnetic field and surface functionalization for reaching synergistic effects in cellular labeling by magnetic core–shell nanospheres†
Tina
Gulin-Sarfraz
a,
Jixi
Zhang
ab,
Diti
Desai
a,
Jarmo
Teuho
c,
Jawad
Sarfraz
a,
Hua
Jiang
d,
Chunfu
Zhang
b,
Cecilia
Sahlgren
ef,
Mika
Lindén
g,
Hongchen
Gu
b and
Jessica M.
Rosenholm
*a
aLaboratory for Physical Chemistry, Åbo Akademi University, Porthansgatan 3, 20500 Turku, Finland. E-mail: jerosenh@abo.fi; Fax: +358 (0)2233 0228; Tel: +358 (0)2215 4253
bMed-X Research Institute and School of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai, P.R. China
cTurku PET Centre, Turku University Hospital, Turku, Finland
dDepartment of Applied Physics, Aalto University, Espoo, Finland
eTurku Centre of Biotechnology, Åbo Akademi University and University of Turku, Turku, Finland
fEindhoven University of Technology, Eindhoven, The Netherlands
gInorganic Chemistry II, University of Ulm, Ulm, Germany
First published on 6th August 2014
Abstract
Aimed at utilizing high-magnetization nanospheres for magnetic field-enhanced cellular labeling, core–shell structured sandwich-like magnetic mesoporous silica nanospheres were developed. While the magnetite cluster core can provide a high magnetic response for overcoming Brownian motion in cell culture media, the layered silica shell facilitates an efficient fluorescent dye labeling. However, the problem of particle aggregation in cell media, which is strongly enhanced under a magnetic field, significantly impeded the uptake by cells, resulting in difficulties in the precise analysis of the degree of particle internalization by fluorescence-based techniques (flow cytometry and confocal microscopy). To overcome this, reflection-based assessment was employed. Further, emphasis was put on utilizing the unique role of surface-hyperbranched polyethylenimine (PEI) in efficient prevention of particle aggregation prior to cell internalization in the presence of an external magnetic field. The interparticle attraction forces originating from magnetic dipole–dipole interactions are hereby balanced by the steric and electrostatic repulsion forces provided by the PEI functionalization, which leads to dispersed nanospheres in cell culture media during the magnetic-field induced cell labeling. As a consequence, PEI functionalization and the presence of the magnetic field synergistically enhanced the efficiency of MRI-fluorescence dual-mode labeling for cellular tracking.
Introduction
Numerous biomedical applications have emerged for superparamagnetic iron oxide nanoparticles (SPIONs),1–6 as they can be easily manipulated by a magnetic field and show great promise for imaging,7,8 magnetic separation,9 magnetic targeting and drug delivery.10–12 Especially for magnetic resonance imaging (MRI), these particles have already made a significant impact,13 where labeling of stem cells is a promising application to noninvasively monitor the distribution and fate of transplanted stem cells.14–16 After the intracellular uptake, SPIONs are metabolized in the lysosomes into a soluble form of iron, that becomes part of the normal iron reservoir in the cells.17,18 Magnetite nanoparticles are commercially available as clinical contrast agents, approved for MR imaging of bowel and liver. The main contrast-enhancing effect of SPIONs originates from the modulation of the transverse relaxation time weighted (T2*) relaxation and thus MR imaging is usually performed using T2/T2* weighted sequences.17 Even if MRI has a lower sensitivity compared to optoacoustic and fluorescence methods, the advantage of MRI is that it does not suffer from penetration depth issues, and is thus suitable for imaging in large animals and humans.19It has been demonstrated that an external magnetic field can enhance the cellular uptake of magnetite nanocrystals.20–24 However, mostly smaller magnetite particles with sizes below 100 nm in size have been studied. It is conceivable that larger particles (crystal aggregates), with a higher total magnetic moment and thus a stronger magnetic separation force in the presence of a magnetic field, could overcome the Brownian motion more efficiently for an improved affinity towards the cell membrane. Clustering of superparamagnetic particles may also provide significantly enhanced relaxivity in comparison to individual magnetite nanoparticles.25,26 However, the application of a magnetic field to suspensions of magnetic particles, particularly those with relatively large particle sizes, can result in the generation of linear aggregates, or chains.27 The applied magnetic field aligns the magnetic moments of the nanoparticles, and the local dipolar fields they generate cause an attraction between nearby particles leading to the generation of chains or linear aggregates of particles aligned along the applied field direction.28 It has also been reported that the system attains an equilibrium state, depending on the local conditions of the particle suspensions (the strength of the magnetic field, the size and polydispersity of the particles, etc.), after a transient period of chain formation.29 The magnetic interaction is what accounts for the inherent instability of the particle system without surface modification and, consequently, its rapid aggregation characteristics.
In order to increase the biocompatibility and biofunctionality of magnetite particles, different methods have been used to coat the particles. The resulting core–shell particles are highly functional nanoparticles with distinct properties arising from the core and the shell layers. Silica is the most widely used coating for inorganic nanoparticles,30 since it is biocompatible, allows for easy further functionalization, and protects the magnetite against oxidation by an acidic environment, and thus may improve the shelf-life of the magnetic cores.31 If the cores are coated with mesoporous silica, the porous shell can also be utilized for incorporation of drugs, to obtain a so-called theranostic probe.32 In addition to drugs, also fluorescent entities may be attached/loaded for multimodal imaging, creating two-in-one fluorescent-magnetic nanocomposites.33 However, a problem when designing these kinds of nanocomposites is the risk of quenching of the fluorophore by the magnetic core, since the magnetite strongly adsorbs the transmitted light.34 Here, a silica coating can thus also be utilized as an effective barrier between the magnetic core and the fluorophores, which may also be controlled by the thickness of the silica shell.
In this study, the cell labeling efficiency of magnetite-silica core–shell nanospheres has been investigated. Core–shell nanospheres with a 120 nm magnetite cluster core and additional non-porous and porous silica layers have been produced, and further studied for magnetically enhanced cellular uptake. The importance of the non-porous silica shell as a barrier between the magnetic core and fluorophore has also been demonstrated. Parallel to the effect of an external magnetic field on cellular labeling efficiency, surface PEI functionalization of the core–shell nanospheres was also considered for aiding the cellular uptake. In addition to this, limitations and shortcomings related to measurements based on fluorescence were observed due to the influence of particle aggregation, especially in the presence of a magnetic field. Thus, reflection-based techniques for determining cellular internalization were studied, as a means to overcome the problems associated with fluorescence. Noteworthy, the actual situation of cellular uptake, where PEI functionalization greatly enhanced cell internalization by protecting particles from aggregation, was explored. Finally, the difference in cellular uptake efficiency could also be discerned using MR-imaging of labelled cells, simultaneously demonstrating the suitability of the produced core–shell nanospheres as cellular labels for MRI-based cellular tracking.
Results and discussion
Design of magnetic core–shell nanospheres for cellular labeling
To facilitate an efficient magnetically-aided nanoparticulate cellular labeling system, core–shell nanospheres with a 120 nm magnetite cluster core and additional non-porous and porous silica shells, Mag@nSiO2@mSiO2, were produced. The size of the magnetic core was chosen both with respect to an effective magnetic-induced enhancement of cellular uptake, due to the increase in magnetization with increased cluster core size; whereas in order to obtain a high contrast in MRI, since it has been demonstrated that a too large core size (≫100 nm) decreases the transverse relaxivity (r2).8 Thus, the size of the magnetic core plays a significant role and has to be chosen according to the requirements for the specific application.Scanning electron microscopy (SEM) image of Mag@nSiO2@mSiO2, as shown in Fig. 1a, reveals an average diameter of almost 300 nm. Typical transmission electron microscopy (TEM) image of Mag@nSiO2@mSiO2 (Fig. 1b) indicates that the nanospheres exhibit a layered sandwich-like core–shell structure. The thickness of the nonporous and mesoporous silica shell is about 60 nm and 30 nm respectively (ESI Fig. S1†). A typical nitrogen sorption isotherm measured for Mag@nSiO2@mSiO2 indicates a type-IV isotherm for mesoporous materials and a pore size distribution with a peak value of 7.3 nm (Fig. 1c). The BET surface area was determined to be 89 m2 g−1 and the pore volume to 0.19 cm3 g−1.
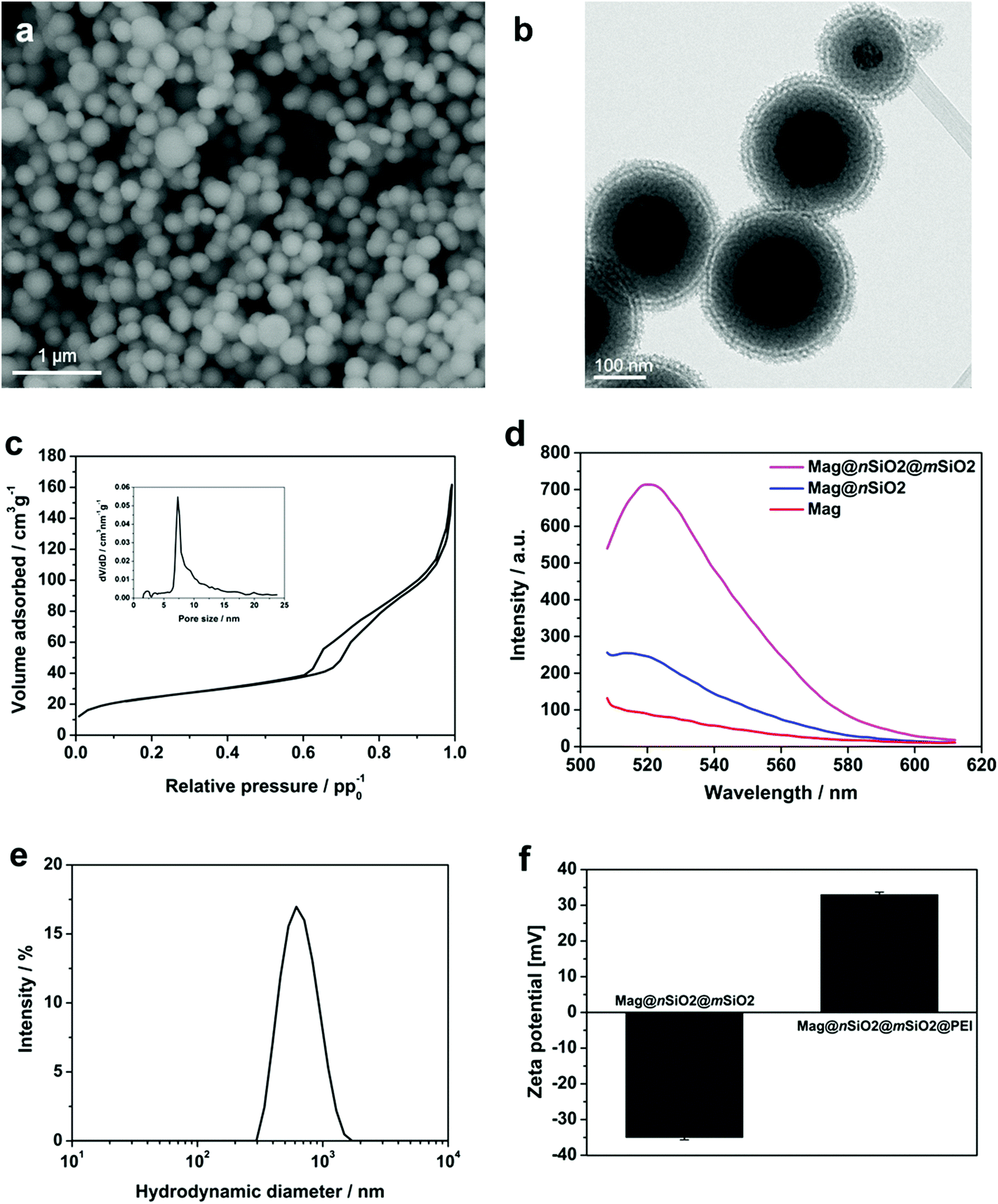 | ||
| Fig. 1 Characterization of the magnetic core–shell nanospheres. (a) SEM image of the Mag@nSiO2@mSiO2 nanospheres, revealing an average size of approximately 300 nm. (b) TEM image showing the morphology and structure of the magnetic cores coated with non-porous and porous silica layers, Mag@nSiO2@mSiO2. (c) Nitrogen sorption isotherm of Mag@nSiO2@mSiO2 nanospheres, with corresponding pore size distribution as determined by the DFT method. The peak at 7 nm corresponds to the average pore diameter. (d) Fluorescence intensities, of similarly FITC-labeled magnetic core particles or magnetic core–shell particles with increasing number of silica layers, were measured in HEPES buffer (pH 7.2) between 510 and 610 nm using 488 nm excitation. The importance of the thicker non-porous silica barrier, towards the magnetite core, on the fluorescence intensity was demonstrated. (e) Hydrodynamic size distribution of the Mag@nSiO2@mSiO2 nanospheres in H2O. The peak is centered at 550 nm with a PdI of 0.15. (f) Zeta potential of Mag@nSiO2@mSiO2 and Mag@nSiO2@mSiO2@PEI nanospheres measured in HEPES buffer, affirms a successful PEI-functionalization with a high positive charge for the PEI-functionalized particles and a negative charge for the non-functionalized particles. | ||
The non-porous silica shell was first deposited on the magnetite cores in order to provide a protective layer between the cores and the fluorophores subsequently labeled on the silica surface, considering the possibility of fluorescence quenching by the magnetic core via an energy transfer process, as well as a strong absorption of the emitted light by the iron oxide particles.34 To demonstrate the role of this protective silica shell, the fluorescence intensity of magnetite cores before and after the coating of the 60 nm thick nonporous layer was compared (Fig. 1d). The Mag and Mag@nSiO2 nanospheres were labeled with same amount of FITC with respect to the mass of the magnetite core. The fluorescence was measured at a particle concentration of 0.5 mg ml−1 (with respect to the magnetite core) in HEPES buffer (pH 7.2) in order to avoid self-quenching from a too high concentration of particles.35 The pure magnetite cores (Mag) were already coated with a very thin silica shell in order to protect the core and keep the core surface hydrophilic, but this thin layer was not sufficient for protecting the fluorophores from quenching by the magnetic cores, resulting in a low fluorescence intensity emission at 520 nm. A 1.5 fold increase in fluorescence intensity could be obtained when increasing the thickness of the nonporous silica shell (Mag@nSiO2). The intensity was further greatly enhanced after the coating of a porous silica shell on the particles (Mag@nSiO2@mSiO2). A porous silica shell helps in keeping the fluorescent molecules more apart due to the large surface area and porous structure, and as a result, the possible self-quenching36 of the fluorophores upon close contact on the particle surface decreases drastically which promotes the high increase in intensity.
The particles were fully dispersible in water (Fig. 1e), with a hydrodynamic size peak centered at 550 nm (z-average) with a PdI of 0.15. The discrepancy between the size of the dried particles measured from SEM images and the hydrodynamic diameter measured by DLS is a known phenomenon due to drawbacks with the DLS technique which can be ascribed to large scattering and absorbance of the particles which may give rise to errors in the size calculation.37–41 However, given the high quality peak and low polydispersity indexes (PdI) we can conclude that the particles were readily dispersible in aqueous solvent after all production steps, whereas the particle sizes are more accurately derived from electron microscopy and thus concluded to be 300 nm on average.
In order to compare the effect of surface functionalization on the cellular labeling efficiency of the core–shell nanospheres, the produced core–shell nanospheres were amino-functionalized with a surface-grafted hyperbranched poly(ethylene imine) (PEI) layer, as this kind of surface coating has shown to be especially facilitative in promoting cellular uptake of silica particles.35,42–44 Electrokinetic measurements (zeta potential) confirmed successful PEI-functionalization (Fig. 1f). The PEI-functionalized nanospheres, Mag@nSiO2@mSiO2@PEI, obtained a high positive charge (+32 mV), while the plain core–shell nanospheres, Mag@nSiO2@mSiO2, presented a typical negative surface charge for silica surfaces (−35 mV). Notably, the employed type of thin porous coating can significantly enhance the degree of surface functionalization, which we have also shown previously.45 FITC-labeling of the particles did not alter the zeta potential of the particles noticeably (ESI Fig. S2†).
Role of surface functionalization and magnetic field on cellular uptake
The cytocompatibility of the produced Mag@nSiO2@mSiO2 and Mag@nSiO2@mSiO2@PEI nanospheres was evaluated by the WST-1 assay at different concentrations of particles in HeLa (human cervical cancer) cells (Fig. 2). No obvious negative effect on cell viability could be discerned at the studied concentrations, regardless if the particles were surface-functionalized or not.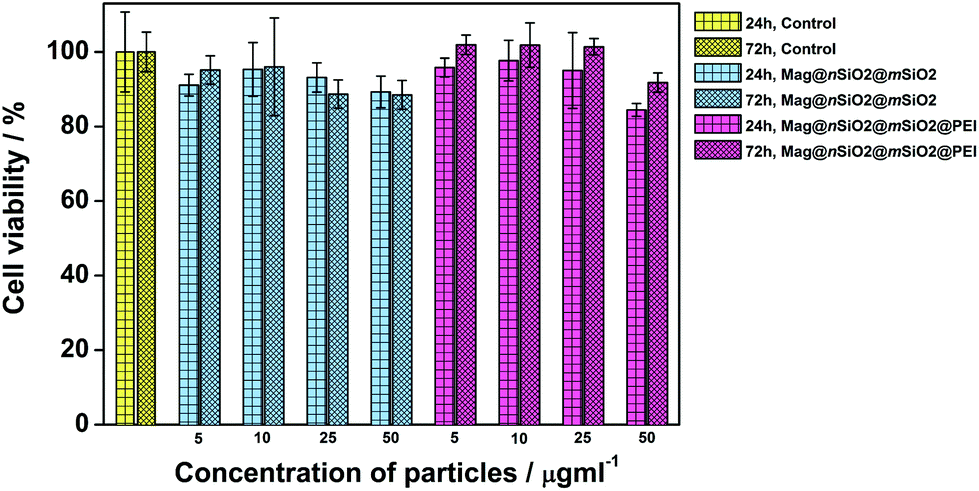 | ||
| Fig. 2 Evaluation of the cytocompatibility of the particles. Cell viability measurement of HeLa cells, incubated with different concentrations of Mag@nSiO2@mSiO2 and Mag@nSiO2@mSiO2@PEI particles, and determined by WST-1 assay revealed sufficient cytocompatibility. | ||
Due to the large size of the magnetic cores and a high saturation magnetization (ESI Fig. S3†), the Mag@nSiO2@mSiO2 nanospheres can be utilized for efficient magnetic field-aided cellular labeling (Fig. 3). Thus, the influence of a magnetic field on the cellular uptake, during a short incubation time of only 1 h, was studied for both plain and PEI-functionalized particles by flow cytometry. HeLa cells were incubated with particles for one hour while subject to an external magnetic field (0.3 T), and subsequently the extracellular fluorescence was quenched by trypan blue.46 The result is presented in Fig. 4a, where ‘internalization’ corresponds to the fluorescence intensity (FL-1) per cell multiplied with the fraction of positive cells. The graph shows apparent higher uptake of the plain nanospheres Mag@nSiO2@mSiO2 (III, +magnet IV) than the positively charged functionalized nanospheres Mag@nSiO2@mSiO2@PEI (I, +magnet II), which is contradictory to what was expected, since it is well known that positively charged silica particles are more extensively taken up by HeLa cells.42 This conflicting result was then further investigated by microscopy (Fig. 4b), without extracellular quenching, to study how the nanospheres “behave” on the cell surface. These images clearly show that the plain core–shell nanospheres are aggregated on the cell surface, and form even larger aggregates when a magnetic field is applied. On the contrary, the positively charged PEI-functionalized nanospheres are well dispersed on the cells, also when the magnetic field is applied, which also readily enhances the cellular uptake, as seen from Fig. 4a. Since the plain nanospheres form large aggregates on the surface of the cells, we believe that these aggregates cannot be effectively quenched by trypan blue, and false high fluorescence intensity, from nearly 100% of the cells, is detected by flow cytometry.
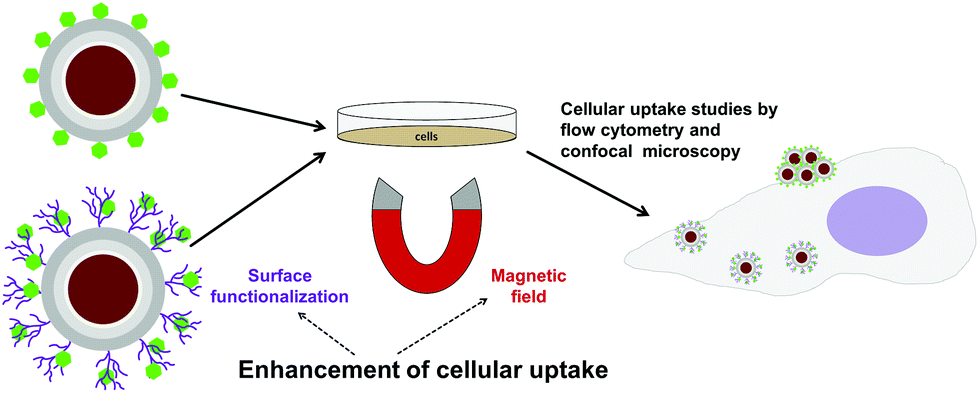 | ||
| Fig. 3 Schematic illustration of the study. The effect of surface functionalization and an external magnetic field on cellular labeling of magnetic core–shell particles was studied. The cellular uptake of the surface functionalized particles could readily be enhanced by a magnetic field, while the non-functionalized particles aggregated on the cell surface which in turn gave rise to false positive fluorescence based signal when measuring the uptake. | ||
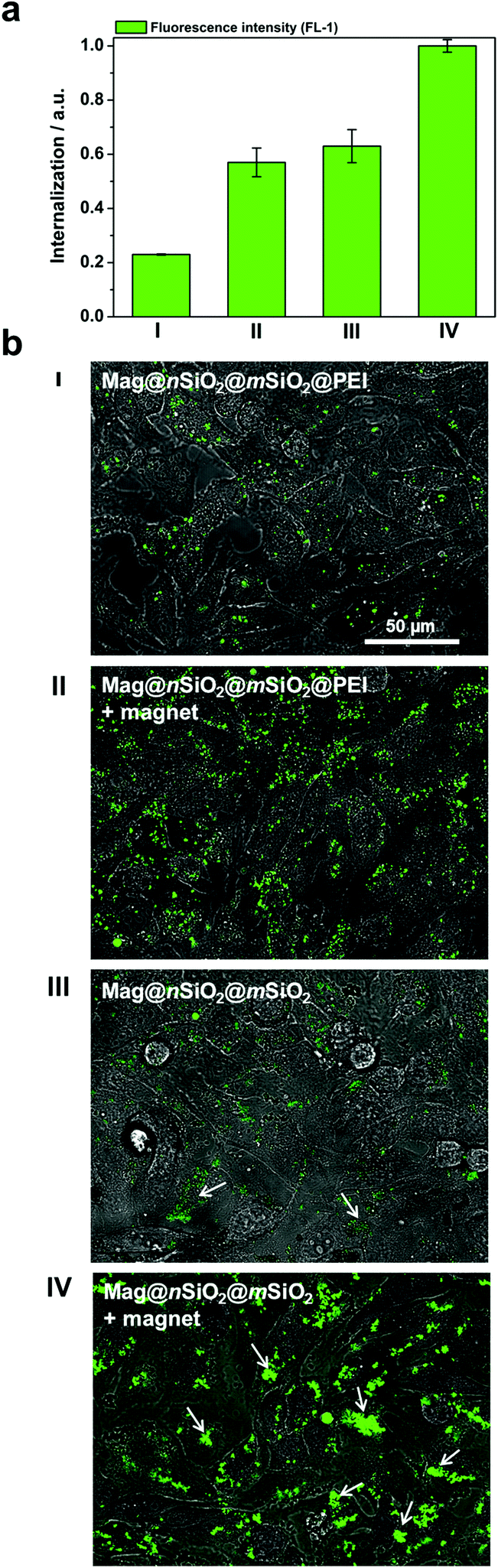 | ||
| Fig. 4 Cellular uptake of particles, based on fluorescence measurements, showing false high uptake due to aggregation and non-efficient quenching of extracellular fluorescence. (a) Cellular uptake of Mag@nSiO2@mSiO2 (III, +magnet IV) and Mag@nSiO2@mSiO2@PEI (I, +magnet II) nanospheres, as determined by flow cytometry. The cells were incubated with 10 μg ml−1 particles for 1 h, with or without an addition of a magnetic field. The extracellular fluorescence was quenched by trypan blue. An addition of a magnetic field can greatly enhance the uptake of the particles, but contradictory to what was expected, higher uptake can be seen for the negatively charged particles. Denotation of the samples are shown in (b). (b) Microscopy images of corresponding samples as presented in (a). No extracellular quenching was used in order to show the distribution of particles on the cells. These images clearly show aggregation of the negatively charged particles. We believe that these aggregates cannot be efficiently quenched and give rise to a high fluorescence signal in flow cytometry. | ||
It has been reported that the magnetic field overcomes the Brownian motion of the particles resulting in their separation from the suspension and an accelerated sedimentation of magnetic iron oxide onto the cell membrane.47 The employment of larger magnetic particles rather than small iron oxide nanocrystals would therefore increase the collision frequency between the particles and the cell membrane, resulting an enhanced affinity toward the cells for being taken up. However, the typical internalization pathways for nanoparticles are greatly dependent on the actual size of the particles.47 It is conceivable that the aggregation of particles before cell uptake would handicap the uptake and lead to a low labeling efficiency of particles. Hence, it is quite crucial to avoid aggregation during cellular uptake/labeling, especially for our Mag@nSiO2@mSiO2 nanospheres with an average diameter of 300 nm.
In order to investigate the aggregation of the particles under cell culturing conditions, both Mag@nSiO2@mSiO2 and Mag@nSiO2@mSiO2@PEI nanospheres were dispersed in cell media and the autocorrelation function of the particle suspensions was measured and compared by dynamic light scattering (DLS) before and after being placed in a magnetic field (0.3 T) for one hour. Considering the distortion of the hydrodynamic diameter by light adsorption of the magnetic core, the autocorrelation function decay, which is related with particle diffusion rate in solution, is a useful tool to study the aggregation behavior of particles.48–50 Since larger particles diffuse slower by Brownian motion than smaller particles, the correlation function decays at a slower rate. Fig. 5a presents the comparison of typical correlation functions for the particles. As the time at which the correlation starts to decay is an indication of the size of the particles, both the Mag@nSiO2@mSiO2 and Mag@nSiO2@mSiO2@PEI have the same hydrodynamic size in the beginning. After the particles were placed in a magnetic field for one hour, the correlation curve for the non-functionalized Mag@nSiO2@mSiO2 nanospheres is significantly shifted to the right with remarkably longer decay time (τ, 106 μs), compared to the curve for the PEI-functionalized nanospheres (104 μs). This indicates large aggregates of the non-functionalized nanospheres has formed, since the signal will decay more slowly if the particles are large and the correlation will persist for a longer time before decaying. The additional plateau in the decay time range from 104 μs to 106 μs before the correlation reaches zero, further affirms the presence of large aggregates. Fig. 5b shows that the Mag@nSiO2@mSiO2 nanospheres sedimented within a couple of seconds after the same magnetic field was applied, again confirming a rapid aggregation of these nanospheres which, in turn, resulted in a higher magnetization and a faster sedimentation and separation rate by the magnetic field. The sterically stabilized Mag@nSiO2@mSiO2@PEI nanospheres kept stable in the magnetic field for longer time, before slowly starting to sediment towards the magnet. When no magnetic field was applied to the particles, they were both stable and no aggregation and/or sedimentation occurred (ESI Fig. S4†).
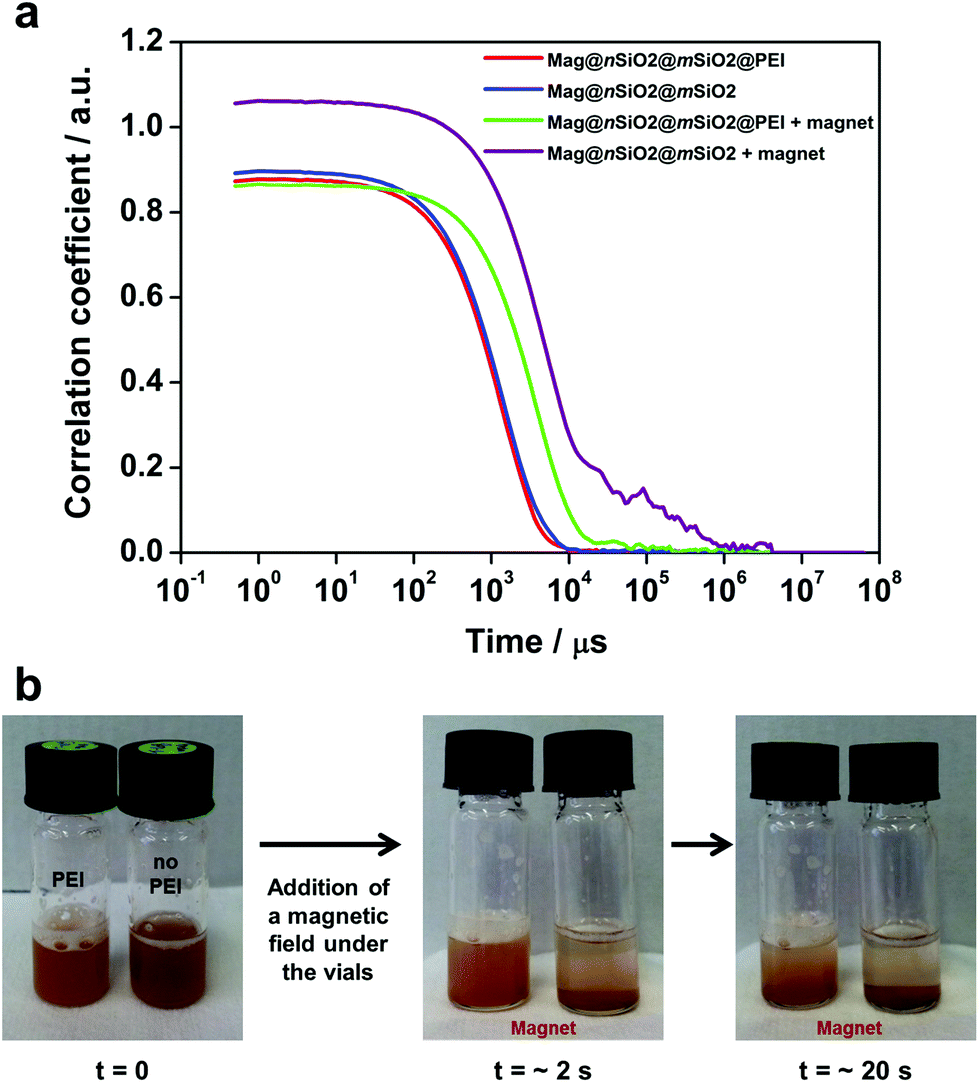 | ||
| Fig. 5 Magnetically induced aggregation of Mag@nSiO2@mSiO2 in cell media as compared to Mag@nSiO2@mSiO2@PEI. (a) Autocorrelation function, measured by DLS for Mag@nSiO2@mSiO2 and Mag@nSiO2@mSiO2@PEI before and after being in contact with a magnetic field for one hour. A clear plateau at higher time points, which indicates large aggregates, could be detected for the Mag@nSiO2@mSiO2 particles. (b) Mag@nSiO2@mSiO2@PEI (left, “PEI”) and Mag@nSiO2@mSiO2 (right, “no PEI”) as freshly dispersed in cell media and after being in contact with a magnetic field for 2 s and 20 s, respectively. More rapid sedimentation can be seen for the Mag@nSiO2@mSiO2 particles due to magnetically induced aggregation to larger clusters with, as a result, faster sedimentation. | ||
Recent experiments show that the electrostatic and steric stabilization strongly influence the aggregation behavior under a magnetic field.28 The electrostatic and steric forces have to be substantial enough to overcome the attractive forces, such as van der Waals and magnetic interactions, to maintain stability.51 Magnetic particles can form chains or linear aggregates,27–29,52 as well as thicker lateral aggregates53 aligned along the applied magnetic field. The magnetically induced aggregation phenomena have been extensively studied both experimentally52 and theoretically,53 where the magnetic interaction has been included into the DLVO theory. It has been reported that the mechanism underlying fast magnetophoretic separation times is reversible magnetically induced aggregation of magnetic particles, taking advantage of the magnetic gradients generated by the particles themselves.52 Thus, the phenomenon is also dependent on the particle concentration.52 The particle size has also been shown to have an effect on the overall stability of dispersions.51
Poly(ethylene imine)-functionalized silica particles, where the polymer is grown by surface hyperbranching polymerization have high suspension stability under biologically relevant conditions.35 Thus, the chemical environment, in this study the hyperbranched PEI layer, acts to slow down and/or prevent aggregation of the nanospheres also under the influence of a magnetic field as was seen in Fig. 5.
We then decided to revisit the fluorescence-based methods for evaluation of the cellular uptake. It is well known that there is a considerable number of problems to be taken into account when following internalized particles based on fluorescence techniques, especially when the extracellular fluorescence signal cannot be completely quenched. However, Stringer et al.54 established a semi-quantitative flow cytometric method, by measuring the change in side scatter (SSC) intensities of alveolar macrophages, to study the uptake of actual environmental particulates without addition of fluorescent molecules. Other research groups55,56 have later suggested that SSC-based flow cytometry measurements might be used as a convenient and fast tool to evaluate the cellular uptake of nanoparticles. Busch et al.57 have compared changes in cellular granularity between different cell and particle types and concluded that the differences were greater between the different particles than between the cell cultures.
Thus, to further study how the particle aggregates affect the results of the cell-uptake measurements, the evaluation of cellular uptake was carried out based on scattering intensities from the cells (forward scattering, FSC, and side scattering, SSC) in flow cytometry. FSC and SSC parameters indicate the size and the intracellular density (granularity) of the cells, respectively, hence FSC additionally elucidate the viability of the cells.55 The cells did not show any change in the FSC channel (ESI Fig. S5†), indicating that the particle uptake did not affect the viability of the cells. Fig. 6a presents the cellular internalization of particles, where internalization corresponds to the fraction of positive cells, in terms of reflection (increased granularity), multiplied with the mean reflectance per cell. Lower uptake of the plain Mag@nSiO2@mSiO2 nanospheres was observed by SSC analysis, regardless of whether the magnetic field was applied or not. In comparison, an approximately 5 times higher uptake of Mag@nSiO2@mSiO2@PEI nanospheres was found and the internalization was further increased by a factor of ∼2 when a magnetic field was applied.
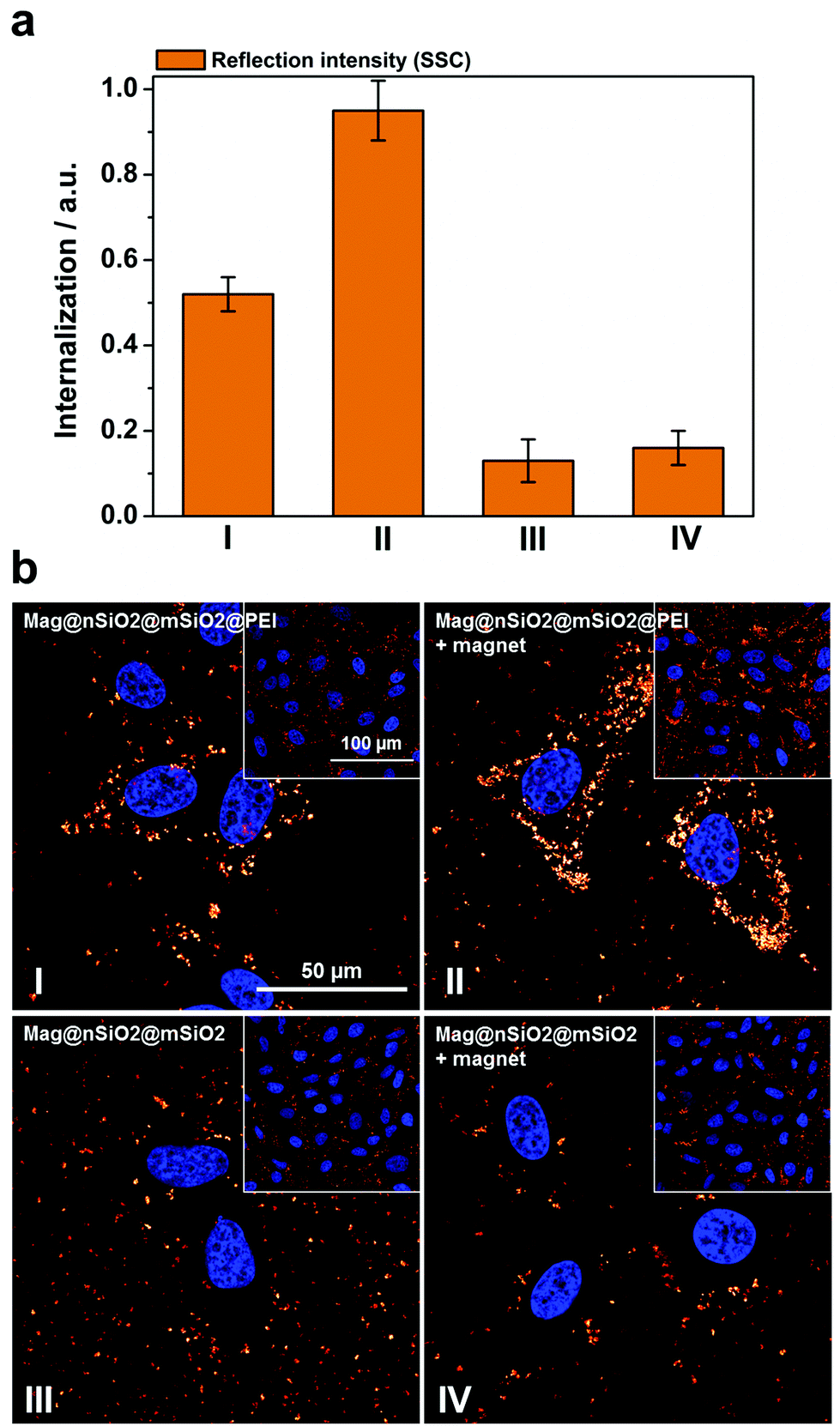 | ||
| Fig. 6 Cellular uptake of particles by means of reflection as a comparison to the fluorescence-based measurements. The same samples, Mag@nSiO2@mSiO2 (III, +magnet IV) and Mag@nSiO2@mSiO2@PEI (I, +magnet II), as measured by fluorescence and presented in Fig. 4, are here presented in terms of reflection. The cells were incubated with 10 μg ml−1 particles for 1 h, with the addition of a magnetic field for the “+magnet” samples. (a) Flow cytometry data analyzed by side scatter of cells (SSC). Higher uptake of the positively charged particles can be seen here, as was expected. Denotation of the samples are shown in (b). (b) Confocal microscopy images of corresponding samples, as presented in (a), based on reflection from the magnetic particles confirm the flow cytometry data. The cell nuclei were stained with DAPI (blue). | ||
The cellular uptake was also investigated by reflection-based confocal microscopy, after one hour of incubation (Fig. 6b). Since the extracellular reflection signal cannot be quenched, thin confocal slices were imaged with the focus set in the middle of the cell nuclei. As seen from the images, the Mag@nSiO2@mSiO2 nanospheres are mostly distributed outside the cells, and no significant difference can be found when a magnetic field has been applied. The positively charged Mag@nSiO2@mSiO2@PEI nanospheres, on the other hand, are much more extensively taken up by the cells, and the particles have accumulated close to the cell nuclei. Moreover, a significantly increased reflection signal from the inside of the cells was found in the presence of the magnetic field. Hence, these results are in good agreement with SSC-based flow cytometry data. Fig. 7 shows the same series of samples in reflection-based confocal microscopy after a longer incubation time (3 h). Noteworthy, the difference in cellular uptake between the functionalized and the plain nanospheres is even more pronounced, as can be seen from the greatly enhanced intracellular reflection signal in the case of Mag@nSiO2@mSiO2@PEI.
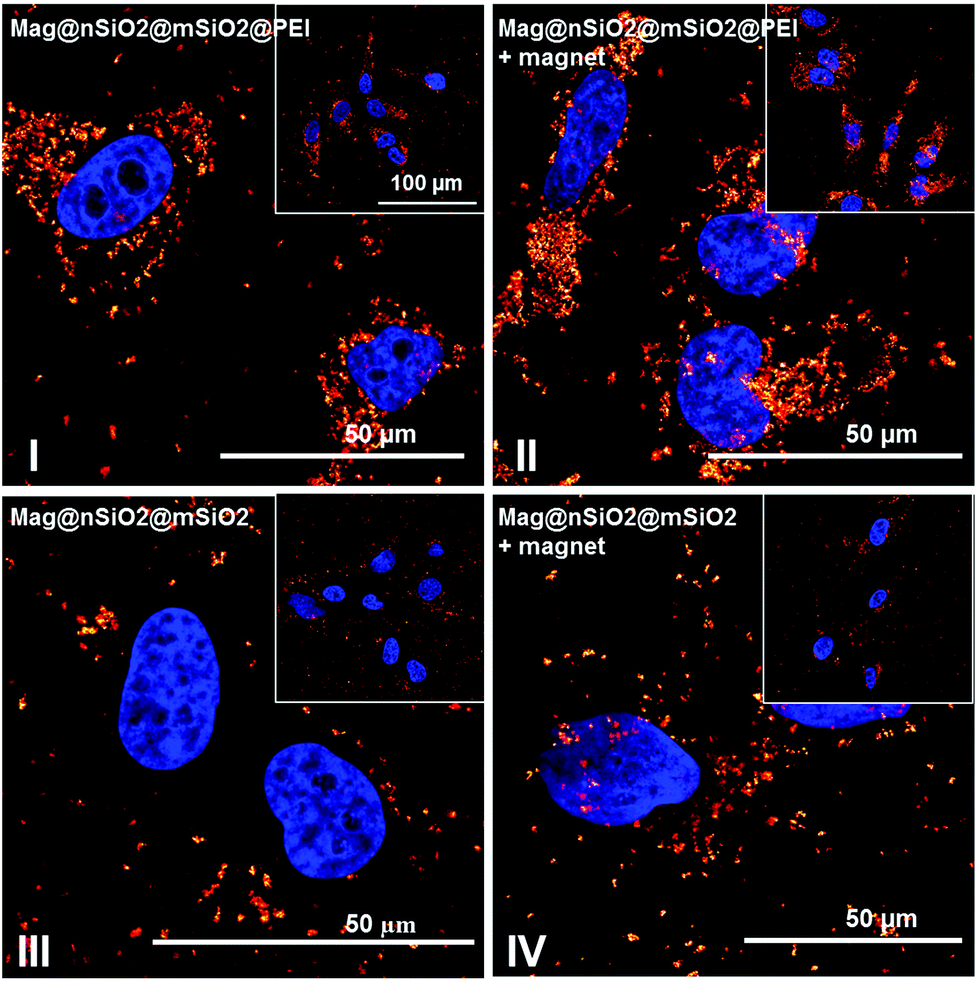 | ||
| Fig. 7 The difference in cellular uptake after 3 h of incubation time. Confocal microscopy images of Mag@nSiO2@mSiO2 and Mag@nSiO2@mSiO2@PEI particles after 3 h of incubation with and without addition of a magnetic field. An even clearer difference in cellular uptake between the positively and negatively charged particles was discerned, with an additional enhancement by the magnetic field. | ||
The time-dependency of the magnetically induced cellular uptake of particles was further investigated with the well-dispersible Mag@nSiO2@mSiO2@PEI nanospheres (Fig. 8). HeLa cells were incubated with particles and analyzed by flow cytometry after various internalization times. The external magnetic field could greatly enhance the cellular uptake especially after short incubation times. Nevertheless, even after three hours of incubation, the effect of the magnetic field was significant. After 24 h of incubation, no difference owing to the magnetic field could be detected, since the amount positive cells even without a magnetic field reached 100%. It is also important to notice, that when no extracellular particle aggregation occurred, measurements based on fluorescence and reflection could be directly compared.
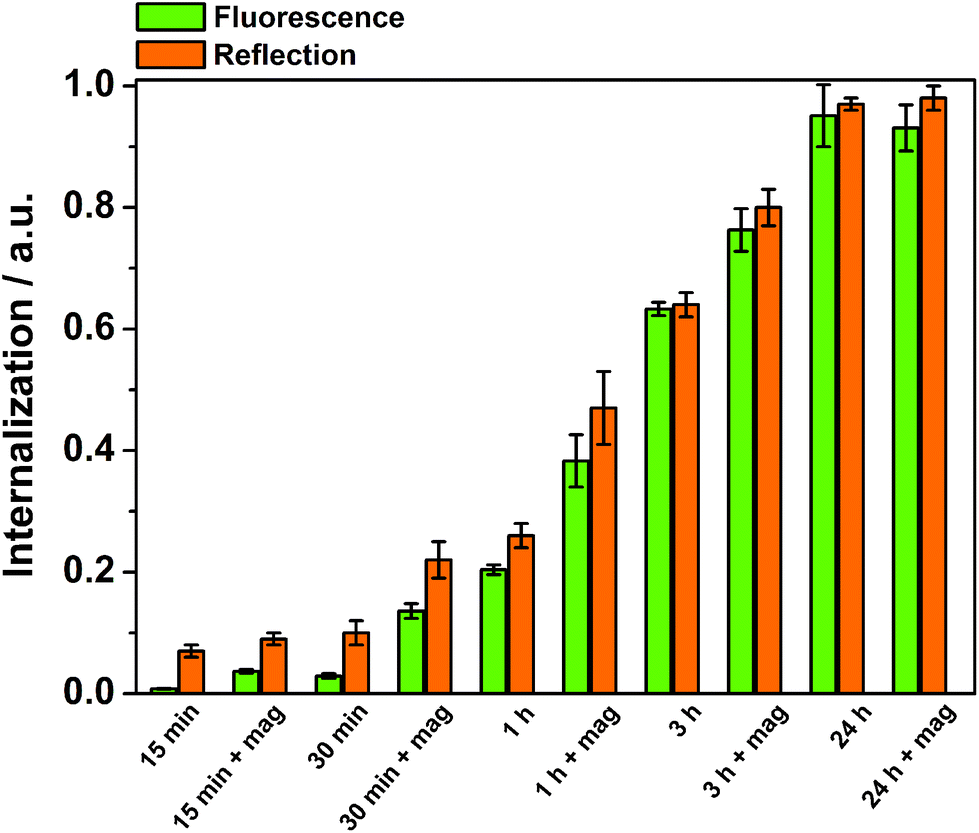 | ||
| Fig. 8 Time-dependency of the magnetically induced cellular uptake of Mag@nSiO2@mSiO2@PEI nanospheres, determined by flow cytometry both based on fluorescence and reflection. A magnetic field can readily enhance the uptake of the particles. The magnetically induced enhancement is more pronounced at short time intervals, but even after 3 h of incubation a clear difference is observed. This simultaneously demonstrates that results obtained from fluorescence and reflection based measurements are comparable if no particle aggregation occur. | ||
On the basis of the results obtained above, it is apparent that magnetic field-induced particle aggregation led to the erroneous determination of cell internalization in the case of Mag@nSiO2@mSiO2. Aided by PEI functionalization, Mag@nSiO2@mSiO2@PEI nanospheres can maintain a good dispersibility in cell culture media for cellular uptake in the presence of a magnetic field. At the same time, we cannot rule out the effect of the PEI coating on the enhanced uptake by its strong interaction with the cell plasma membrane, which has been observed in previous studies.35,42–44 Hence, in the present case, the surface PEI functionalization exhibits a dual functionality for an efficient cellular labeling of the particles.
The efficiency of cellular labeling of the core–shell nanospheres as investigated by magnetic resonance imaging
In order to evaluate the efficiency of the produced Mag@nSiO2@mSiO2@PEI nanospheres as magnetic resonance imaging (MRI) contrast agents, HeLa cells after particle labeling for different time periods with/without the presence of an external magnetic field were measured and compared on a clinical 3 T MRI instrument. T2-weighted phantom images (Fig. 9a) clearly show the T2 shortening effect (signal loss) in the samples where cells were treated and/or labeled with particles at a concentration of 10 μg ml−1. This demonstrates that the Mag@nSiO2@mSiO2@PEI nanospheres were efficiently taken up by HeLa cells and served as a contrast agent for the labeled cells being visualized by MRI. Moreover, the extent of T2 shortening shows the same trend of dependency on the treatment time and the magnetic field, as already demonstrated with flow cytometry. This is reflected in the measured 1/T2 values as shown in Fig. 9b. A 2.5-fold increase in 1/T2 value, for the labeled sample compared to the control sample, is seen after 30 min. When a magnetic field was applied, 1/T2 was further increased by more than 2-fold as compared with the sample labeled in the absence of the magnetic field, which in total gives almost a 6-fold increase compared to the control cells, indicative of the magnetic enhanced uptake of particles. After further increase of 1/T2 after 1 h of labeling to almost 7-fold compared to control cells, no significant further enhancement caused by the magnetic field can be observed at the 3 h time-point, revealing a saturated enhancement of MRI signal at a concentration of 10 μg ml−1. An incubation time of 24 h was most often presented in similar MRI studies where cell labeling were involved.31,58,59 This further confirms the significant advantage of a magnetic field for efficiently enhancing the cellular uptake and decreasing the incubation time in our Mag@nSiO2@mSiO2@PEI system.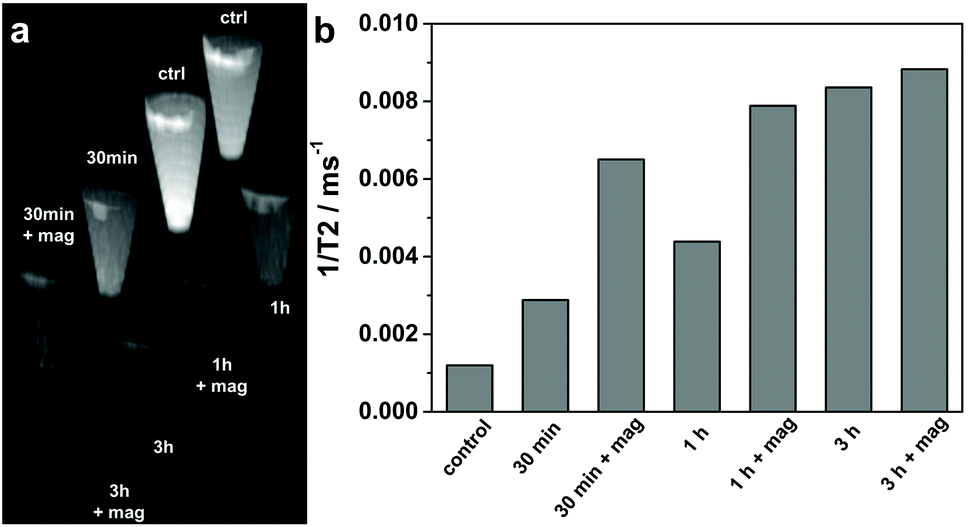 | ||
| Fig. 9 MR imaging comparison of labeled cells as a function of treatment time with/without the presence of an external magnetic field. (a) MRI phantom images of cells labeled with 10 μg ml−1 Mag@nSiO2@mSiO2@PEI nanospheres for different time intervals with and without magnetically-enhanced uptake. The same concentration of HeLa cells were used (1.5 × 106 cells per tube) for comparison. (b) The 1/T2 values, derived from the corresponding images, as a function of various labeling time points. The trend, which these results show, confirms the positive effect of the magnetic field on the cellular uptake. When a magnetic field was applied for 30 min, an almost 6-fold increase in 1/T2 was found compared to the control cells. | ||
Experimental
Reagents and materials
Aziridine (98%) was purchased from Menadiona. Tetraethyl orthosilicate (TEOS, ≥98%) and toluene (99.8%) were obtained from Fluka. 2-Propanol (99.5%), N,N-dimethylformamide (DMF, ≥99.9%), ammonium hydroxide (NH3, 33 wt%) and polyoxyethylene(20)-sorbitan monolaurate (Tween 20) were purchased from Sigma-Aldrich. Sodium chloride (NaCl, 99.7%) was obtained from J. T. Baker and absolute ethanol (99%) from Altia. Fluorescein isothiocyanate (FITC, 90%), 3-aminopropyl-triethoxysilane (APTES, 99 wt%) and poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (P123) were obtained from Aldrich. Acetone (≥99.8%) and acetic acid (CH3COOH, ≥99.8%) were purchased from Merck. All chemicals used for the study were of analytical grade and Milli-Q water (18.2 MΩ cm) was used throughout the study.For the in vitro studies, HeLa (cervical carcinoma) cells were seeded in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. DMEM, Dulbecco's phosphate buffered saline (PBS) and Mowiol® were obtained from Sigma. Paraformaldehyde (PFA) and trypsin (0.25% trypsin, 0.02% K-EDTA) were purchased from Fluka, trypan blue (TB) from Sigma-Aldrich, Vectashield® containing DAPI from Vector, and WST-1 reagent from Roche Applied Science.
Synthesis of magnetic core–shell nanospheres, Mag@nSiO2@mSiO2
Magnetite cores (Mag) with an average diameter of 120 nm and a thin nonporous silica shell synthesized based on a well-established approach,60 were provided by Shanghai AllrunNano Science & Technology Co. Then, a thicker non-porous silica coating was deposited on the magnetite cores. Typically, 40 mg magnetite cores was separated by a magnet, washed with water, and redispersed into 20 ml of Tween 20 solution (0.5 wt%) by sonication for 20 min. Afterwards, 40 ml 2-propanol was added under mechanical stirring at 350 rpm. 0.8 ml 33 wt% NH3 and 0.3 ml TEOS were added and the stirring continued for 48 h. The particles were separated with centrifugation and washed 3 times with ethanol. The as-obtained product was denoted as Mag@nSiO2.The procedure described by Rosenholm et al.61 was used with slight modifications to coat the particles with a large-pore mesoporous silica shell. Typically, 0.05 g P123 and 0.29 g NaCl were dissolved in a mixture of 40 ml water and 16 ml ethanol by ultrasonicating for 15 min. Subsequently, 25 mg silica coated magnetite was added and the dispersion was left stirring at 350 rpm at 35 °C for 30 min. Then, 0.22 ml TEOS was added and the stirring continued for another 24 h. The template was extracted from the shell by sonication in acetone for 30 min and this step was repeated 3 times. The final product was denoted as Mag@nSiO2@mSiO2.
Amino-functionalization and fluorophore labeling of the core–shell nanospheres
The Mag@nSiO2@mSiO2 nanospheres were PEI-functionalized according to the procedure, described by Rosenholm et al.62 In a typical functionalization procedure 10 mg Mag@nSiO2@mSiO2 were washed with toluene and then dispersed in 20 ml toluene. Afterwards, 1 μl acetic acid and 10 μl aziridine were added successively and the reaction mixture was sonicated at 75 °C for 5 h, after which the mixture was left standing at 75 °C for 20 h. The Mag@nSiO2@mSiO2@PEI nanospheres were finally washed with acetone.Different procedures for FITC-labeling were used depending on the surface groups of the particles. The synthesis of FITC-labeled Mag, Mag@nSiO2, and Mag@nSiO2@mSiO2 was performed by a silane mediated conjugation process. In brief, a FITC-solution containing 4 mg FITC, 2.4 ml ethanol and 10 μl APTES was first prepared. This mixture was stirred for 2 h under inert gas. Subsequently, 10 mg particles were dispersed in 10 ml toluene and 0.5 ml FITC-solution was added and the mixture was left stirring for 24 h. The particles were separated by centrifugation and washed with toluene and ethanol.
For the FITC labeling on the Mag@nSiO2@mSiO2@PEI, the preparation was carried out by conjugation of FITC to the amine groups in the surface-polymerized PEI. Typically, 5 mg particles were suspended in 1.5 ml DMF, and 25 μl FITC dissolved in DMF (1 mg ml−1) was added. The reaction continued under stirring for 4 h after which the particles were separated by centrifugation and washed with DMF and ethanol.
Characterization methods
The size distribution of the nanospheres was determined by scanning electron microscopy (SEM) using a Zeiss DSM 962 at 5 kV. High-resolution transmission electron microscopy (TEM) was used to determine the morphology of the nanospheres with an aberration-corrected JEOL-2200FS microscope, operated at 200 kV. Zeta potential measurements were carried out to determine the net surface charge of the core–shell nanospheres before and after PEI-functionalization. Dynamic light scattering, DLS, was used for measuring the dispersibility of the particles. For zeta potential and DLS measurements the instrument Malvern ZetasizerNano ZS was used. The intensity of the fluorescent samples was measured with a PerkinElmer LS50B luminescence spectrometer.Cell viability measurements
HeLa cells were plated at a density of 5000 cells per well into the 96-well plates and incubated overnight. After that, the cell medium was removed and replaced with 100 μl medium containing different concentrations of either Mag@nSiO2@mSiO2 or Mag@nSiO2@mSiO2@PEI (5, 10, 25, 50 μg ml−1). Four replicates were used at each concentration and only cell media was added in the control group. After incubation for 24–72 h, 10 μl of WST-1 reagent was added into the plate and incubated for 90 min (standard incubation time) at 37 °C in 5% CO2. The absorbance of the colored formazan was measured at 430 nm wavelength using TECAN ULTRA Microplate Reader (Tecan Group Ltd). To avoid any interference of the dark color of the magnetite particles with the spectrophotometry readings, the net readings were corrected with readings of the same amount of particle-labelled cells which had not been incubated with the WST-1 reagent. The relative cell viability percentage in each group was calculated by comparison to the control group.Cellular uptake determination by flow cytometry
FITC-labeled nanospheres were suspended in cell media at a concentration of 10 μg ml−1, and sonicated for 15 min. The particle suspension was added to the cells and incubated for different time intervals with or without a magnet placed under the cell plate. Fitted magnets, with a magnetic field strength of 0.3 T, were used under the cell plates.21 After incubation the cells were washed with PBS, detached by trypsinization, centrifuged down at 2000 rpm for 5 min and again washed with PBS. The extracellular fluorescence was quenched by re-suspending the cells in trypan blue (200 μg ml−1) and incubating them for 10 min at room temperature. After washing once with PBS, the cells were re-suspended in PBS and analyzed with a BD FacsCalibur flow cytometer. The forward scatter and side scatter intensities, indicating the size and intracellular density of the cells, were simultaneous recorded with the mean fluorescence intensity (MFI) of the cells at FL-1 channel (ex 488 nm, em 530/30 nm). The data was analyzed with BD CellQuestPro™ software. The control peak (no particles) was subtracted from the sample peak, whereafter the resulting fluorescence intensity (MFI) value was normalized against the fluorescence intensity for particle suspensions only, and further multiplied with the fraction of positive cells as representation for internalization.Confocal microscopy imaging
For the intracellular imaging, HeLa cells were grown on cover slips and incubated with nanospheres for different time intervals as described above. Particle uptake was stopped by washing off most non-internalized nanospheres with PBS and fixing of the cells with 3% PFA for 15 min at room temperature. After washing with PBS the cells were mounted on microscope slides (VWR) using Mowiol® or Vectashield® containing DAPI. A Zeiss LSM780 confocal microscope, equipped with a 20×/0.5 or 40×/1.2 water immersion objective, was used to record fluorescence (FITC: ex 488 nm, em 500–550 nm; DAPI: ex 405 nm, em 410–500 nm) and reflection (ex 405 nm, em 387–410 nm) images, using 68 μm pinhole size.Magnetic resonance imaging
For magnetic resonance imaging of cells, HeLa cells were incubated with nanospheres as described above. Particle uptake was stopped by washing the cells 3 times with PBS, after which the cells were trypsinized, centrifuged down and washed with PBS again. Cells were then transferred to eppendorf tubes at a concentration of 1.5 × 106 cells per tube and pelleted. Finally each eppendorf tube was filled with 0.5 ml of 3% gelatin and then the samples were left to harden before MRI measurements. MR imaging was performed with a Philips Ingenuity TF 3 T PET/MRI scanner (Philips Medical Systems). The scanner is a hybrid imaging system with Philips time-of-flight GEMINI TF PET and Achieva 3 T X-series MRI system. MR imaging was performed using a dedicated small animal coil (Rat Whole Body Coil, Rapid Biomedical GmbH), with a 2D T2-weighted spin echo (SE) sequence (echo time TE = 15–180 ms, repetition time TR = 1670 ms, 12 echoes, slice thickness 2 mm) for the contrast agent phantoms. The T2 relaxation times were calculated by an exponential fit of the region-of-interest signal amplitudes versus TE.Conclusion
Core–shell structured fluorescent magnetic nanospheres with a magnetite cluster core and additional porous and non-porous silica layers have been produced and studied for magnetically enhanced cellular labeling. The developed core–shell nanospheres were further modified by surface hyperbranching polymerization of poly(ethyleneimine) (PEI), in order to compare the effect of surface functionalization with the magnetic field-induced effect on the cellular labeling efficiency. Magnetically induced aggregation behavior of non-functionalized magnetic nanospheres was demonstrated under cell relevant conditions by DLS and further confirmed under in vitro conditions. The aggregation led to difficulties in correctly assessing the cellular uptake behavior of these magnetic nanospheres by fluorescence-based techniques, which were overcome by a precise determination by using flow cytometry based on reflection analysis instead. A synergistic effect of the magnetic field together with the particle dispersibility improved by PEI functionalization could greatly enhance the cellular internalization of particles, which further provided for a high MRI contrast after a short incubation time. We expect that this Mag@nSiO2@mSiO2@PEI nanosphere platform can serve as a promising two-in-one fluorescent-magnetic tool for bimodal imaging and cellular labeling for related biomedical applications.Acknowledgements
The Graduate School of Åbo Akademi University (TG-S), Magnus Ehrnrooth Foundation (JZ), Centre for International Mobility CIMO (DD), the European Regional Development Fund in South Finland (JS), TEKES (the Finnish Funding Agency for Technology and Innovation) NAMI (China-Finland Nanotechnology Strategic Mutual Collaboration Initiative) project #19807 (TG-S, ML, JMR) and Academy of Finland project decisions #131034 (CS), #137101/140193/260599/278812 (JMR) are gratefully acknowledged for financial support. The MRI part of the study was conducted within the Finnish Center of Excellence in Molecular Imaging in Cardiovascular and Metabolic Research and strategic Japanese-Finnish research cooperative program on “Application of Medical ICT Devices” supported both by the Academy of Finland, University of Turku, Turku University Hospital and Åbo Akademi University (JT). Helena Saarento is acknowledged for kind help with cell culture preparation. We would also like to acknowledge the assistance by Jouko Sandholm at Cell Imaging Core, Turku Centre of Biotechnology, during confocal microscopy imaging; Markus Peurla from Laboratory of Electron Microscopy University of Turku during the TEM characterization; and the assistance by Jyrki Juhanoja at Top Analytica Oy Ab during the SEM characterization. This work made as well use of the Aalto University Nanomicroscopy Center (Aalto-NMC) premises.Notes and references
- L. H. Reddy, J. L. Arias, J. Nicolas and P. Couvreur, Chem. Rev., 2012, 112, 5818–5878 CrossRef CAS PubMed.
- Y. Tai, L. Wang, G. Yan, J. Gao, H. Yu and L. Zhang, Polym. Int., 2011, 60, 976–994 CrossRef CAS PubMed.
- C. Xu and S. Sun, Adv. Drug Delivery Rev., 2013, 65, 732–743 CrossRef CAS PubMed.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- Q. A. Pankhurst, N. K. T. Thanh, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42, 224001 CrossRef.
- J. R. McCarthy and R. Weissleder, Adv. Drug Delivery Rev., 2008, 60, 1241–1251 CrossRef CAS PubMed.
- F. Xu, C. Cheng, F. Xu, C. Zhang, H. Xu, X. Xie, D. Yin and H. Gu, Nanotechnology, 2009, 20, 405102 CrossRef PubMed.
- F. Xu, C. Cheng, D.-X. Chen and H. Gu, ChemPhysChem, 2012, 13, 336–341 CrossRef CAS PubMed.
- I. Šafařík and M. Šafaříková, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 722, 33–53 CrossRef.
- Wahajuddin and S. Arora, Int. J. Nanomedicine., 2012, 7, 3445–3471 CrossRef CAS PubMed.
- J. d. g. Durán, J. l. Arias, V. Gallardo and A. v. Delgado, J. Pharm. Sci., 2008, 97, 2948–2983 CrossRef PubMed.
- M. Arruebo, R. Fernández-Pacheco, M. R. Ibarra and J. Santamaría, Nano Today, 2007, 2, 22–32 CrossRef.
- Z. R. Stephen, F. M. Kievit and M. Zhang, Mater. Today, 2011, 14, 330–338 CrossRef CAS.
- L. Li, Theranostics, 2013, 3, 595–615 CrossRef PubMed.
- J. M. I. Wiehe, O. Zimmermann, J. Greiner, J. M. Homann, M. Wiesneth, V. Hombach and J. Torzewski, Histol. Histopathol., 2005, 20, 901–906 CAS.
- L. Zhang, Y. Wang, Y. Tang, Z. Jiao, C. Xie, H. Zhang, P. Gu, X. Wei, G.-Y. Yang, H. Gu and C. Zhang, Nanoscale, 2013, 5, 4506–4516 RSC.
- Y.-X. J. Wang, Quant. Imaging Med. Surg., 2011, 1, 35–40 Search PubMed.
- S. P. Foy and V. Labhasetwar, Biomaterials, 2011, 32, 9155–9158 CrossRef CAS PubMed.
- A. Taylor, K. M. Wilson, P. Murray, D. G. Fernig and R. Lévy, Chem. Soc. Rev., 2012, 41, 2707–2717 RSC.
- Q. Liu, J. Zhang, W. Xia and H. Gu, J. Nanosci. Nanotechnol., 2012, 12, 7709–7715 CrossRef CAS PubMed.
- Q. Liu, J. Zhang, W. Xia and H. Gu, Nanoscale, 2012, 4, 3415–3421 RSC.
- C.-A. M. Smith, J. de la Fuente, B. Pelaz, E. P. Furlani, M. Mullin and C. C. Berry, Biomaterials, 2010, 31, 4392–4400 CrossRef CAS PubMed.
- H. W. Child, P. A. Del Pino, J. M. De La Fuente, A. S. Hursthouse, D. Stirling, M. Mullen, G. M. McPhee, C. Nixon, V. Jayawarna and C. C. Berry, ACS Nano, 2011, 5, 7910–7919 CrossRef CAS PubMed.
- S. Chaudhary, C. A. Smith, P. del Pino, J. M. de la Fuente, M. Mullin, A. Hursthouse, D. Stirling and C. C. Berry, Pharmaceuticals, 2013, 6, 204–222 CrossRef CAS PubMed.
- D. Patel, A. Kell, B. Simard, J. Deng, B. Xiang, H.-Y. Lin, M. Gruwel and G. Tian, Biomaterials, 2010, 31, 2866–2873 CrossRef CAS PubMed.
- J. Kim, J. E. Lee, S. H. Lee, J. H. Yu, J. H. Lee, T. G. Park and T. Hyeon, Adv. Mater., 2008, 20, 478–483 CrossRef CAS PubMed.
- J. Faraudo, J. S. Andreu and J. Camacho, Soft Matter, 2013, 9, 6654–6664 RSC.
- S. L. Saville, R. C. Woodward, M. J. House, A. Tokarev, J. Hammers, B. Qi, J. Shaw, M. Saunders, R. R. Varsani, T. G. S. Pierre and O. T. Mefford, Nanoscale, 2013, 5, 2152–2163 RSC.
- J. S. Andreu, J. Camacho and J. Faraudo, Soft Matter, 2011, 7, 2336–2339 RSC.
- A. Guerrero-Martínez, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Mater., 2010, 22, 1182–1195 CrossRef PubMed.
- J. L. Campbell, J. Arora, S. F. Cowell, A. Garg, P. Eu, S. K. Bhargava and V. Bansal, PLoS One, 2011, 6, e21857 CAS.
- J. E. Lee, N. Lee, T. Kim, J. Kim and T. Hyeon, Acc. Chem. Res., 2011, 44, 893–902 CrossRef CAS PubMed.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438–8441 CrossRef CAS PubMed.
- S. A. Corr, Y. P. Rakovich and Y. K. Gun'ko, Nanoscale Res. Lett., 2008, 3, 87–104 CrossRef CAS.
- D. Desai, D. S. Karaman, N. Prabhakar, S. Tadayon, A. Duchanoy, D. M. Toivola, S. Rajput, T. Näreoja and J. M. Rosenholm, Mesoporous Biomater., 2014, 1, 16–43 Search PubMed.
- H. C. Ishikawa-Ankerhold, R. Ankerhold and G. P. C. Drummen, Molecules, 2012, 17, 4047–4132 CrossRef CAS PubMed.
- K. Möller, J. Kobler and T. Bein, Adv. Funct. Mater., 2007, 17, 605–612 CrossRef PubMed.
- R. De Palma, S. Peeters, M. J. Van Bael, H. Van den Rul, K. Bonroy, W. Laureyn, J. Mullens, G. Borghs and G. Maes, Chem. Mater., 2007, 19, 1821–1831 CrossRef CAS.
- A. Dhawan and V. Sharma, Anal. Bioanal. Chem., 2010, 398, 589–605 CrossRef CAS PubMed.
- F. Lu, S.-H. Wu, Y. Hung and C.-Y. Mou, Small, 2009, 5, 1408–1413 CrossRef CAS PubMed.
- S. I. Jenkins, M. R. Pickard, D. N. Furness, H. H. P. Yiu and D. M. Chari, Nanomedicine, 2013, 8, 951–968 CrossRef CAS PubMed.
- D. S. Karaman, D. Desai, R. Senthilkumar, E. M. Johansson, N. Råtts, M. Odén, J. E. Eriksson, C. Sahlgren, D. M. Toivola and J. M. Rosenholm, Nanoscale Res. Lett., 2012, 7, 358 CrossRef PubMed.
- J. Pang, L. Zhao, L. Zhang, Z. Li and Y. Luan, J. Colloid Interface Sci., 2013, 395, 31–39 CrossRef CAS PubMed.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Lindén, ACS Nano, 2008, 3, 197–206 CrossRef PubMed.
- T. Gulin-Sarfraz, J. Sarfraz, D. Ş. Karaman, J. Zhang, C. Oetken-Lindholm, A. Duchanoy, J. M. Rosenholm and D. Abankwa, RSC Adv., 2014, 4, 16429–16437 RSC.
- S. Sahlin, J. Hed and I. Rundquist, J. Immunol. Methods, 1983, 60, 115–124 CrossRef CAS.
- S. Huth, J. Lausier, S. W. Gersting, C. Rudolph, C. Plank, U. Welsch and J. Rosenecker, J. Gene Med., 2004, 6, 923–936 CrossRef CAS PubMed.
- J. Lim, S. P. Yeap, H. X. Che and S. C. Low, Nanoscale Res. Lett., 2013, 8, 381 CrossRef PubMed.
- G. K. Min, M. A. Bevan, D. C. Prieve and G. D. Patterson, Colloids Surf., A, 2002, 202, 9–21 CrossRef CAS.
- B. J. Berne and R. Pecora, Dynamic Light Scattering: With Applications to Chemistry, Biology, and Physics, Courier Dover Publications, 2000 Search PubMed.
- O. T. Mefford, M. L. Vadala, J. D. Goff, M. R. J. Carroll, R. Mejia-Ariza, B. L. Caba, T. G. S. Pierre, R. C. Woodward, R. M. Davis and J. S. Riffle, Langmuir, 2008, 24, 5060–5069 CrossRef CAS PubMed.
- G. De Las Cuevas, J. Faraudo and J. Camacho, J. Phys. Chem. C, 2008, 112, 945–950 CAS.
- J. Faraudo and J. Camacho, Colloid Polym. Sci., 2010, 288, 207–215 CAS.
- B. Stringer, A. Imrich and L. Kobzik, Cytometry, 1995, 20, 23–32 CrossRef CAS PubMed.
- J. Xia, S. Zhang, Y. Zhang, M. Ma, K. Xu, M. Tang and N. Gu, J. Nanosci. Nanotechnol., 2008, 8, 6310–6315 CrossRef CAS PubMed.
- H. Suzuki, T. Toyooka and Y. Ibuki, Environ. Sci. Technol., 2007, 41, 3018–3024 CrossRef CAS.
- W. Busch, S. Bastian, U. Trahorsch, M. Iwe, D. Kühnel, T. Meißner, A. Springer, M. Gelinsky, V. Richter, C. Ikonomidou, A. Potthoff, I. Lehmann and K. Schirmer, J. Nanopart. Res., 2011, 13, 293–310 CrossRef CAS.
- J.-K. Hsiao, M.-F. Tai, Y.-C. Lee, C.-Y. Yang, H.-Y. Wang, H.-M. Liu, J.-S. Fang and S.-T. Chen, J. Magn. Magn. Mater., 2006, 304, e4–e6 CrossRef CAS PubMed.
- S. R. Bhattarai, R. B. Kc, S. Y. Kim, M. Sharma, M. S. Khil, P. H. Hwang, G. H. Chung and H. Y. Kim, J. Nanobiotechnol., 2008, 6, 1–9 CrossRef PubMed.
- H. Xu, L. Cui, N. Tong and H. Gu, J. Am. Chem. Soc., 2006, 128, 15582–15583 CrossRef CAS PubMed.
- J. M. Rosenholm, J. Zhang, W. Sun and H. Gu, Microporous Mesoporous Mater., 2011, 145, 14–20 CrossRef CAS PubMed.
- J. M. Rosenholm, A. Penninkangas and M. Lindén, Chem. Commun., 2006, 3909–3911 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4bm00221k |
| This journal is © The Royal Society of Chemistry 2014 |