
Quantitative study on the antifreeze protein mimetic ice growth inhibition properties of poly(ampholytes) derived from vinyl-based polymers†
Daniel E.
Mitchell
ab,
Mary
Lilliman
a,
Sebastian G.
Spain
c and
Matthew I.
Gibson
 *ad
*ad
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: m.i.gibson@warwick.ac.uk; Fax: +44 (0)2476524112
bMolecular Organisation and Assembly in Cells (MOAC) Doctoral Training Centre, University of Warwick, CV4 7A, UK
cSchool of Pharmacy, University of Nottingham, Nottingham NG7 2RD, UK
dWarwick Medical School, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK
First published on 15th September 2014
Abstract
Antifreeze (glyco) proteins (AF(G)Ps) from the blood of polar fish species are extremely potent ice recrystallization inhibitors (IRI), but are difficult to synthesise or extract from natural sources. Despite this challenge, materials which display IRI are appealing due to their ability to enhance cellular cryopreservation, for applications including regenerative and transplantation medicine. Here, poly(ampholytes), which contain a mixture of cationic and anionic side chains are quantitatively evaluated for their IRI activity. Poly(aminoethyl methacrylate), obtained by RAFT polymerization, is functionalised with succinic anhydride to generate the poly(ampholytes). The charge balance of the side chains is shown to be crucial, with only 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixtures having strong IRI activity, which also scales with molecular weight. This is the first example of a non-hydroxylated synthetic polymer with quantifiable IRI activity and raises questions about the mechanism of IRI, as the polymers have no obvious ice-binding motif. The ampholytic structure is shown to be transferable to carbohydrate-centred polymers with activity retained, but poly(betaines) are shown to be inactive.
:50 mixtures having strong IRI activity, which also scales with molecular weight. This is the first example of a non-hydroxylated synthetic polymer with quantifiable IRI activity and raises questions about the mechanism of IRI, as the polymers have no obvious ice-binding motif. The ampholytic structure is shown to be transferable to carbohydrate-centred polymers with activity retained, but poly(betaines) are shown to be inactive.
Introduction
Since their discovery in the serum of Arctic fish species, antifreeze proteins (AFPs) and antifreeze (glyco)proteins (AF(G)Ps) have attracted significant attention due to their unique ice-interacting properties and their potential applications in cryopreservation, anti-icing surfaces, food storage and more.1–3 AF(G)Ps have three key macroscopic properties;4,5 (i) thermal hysteresis (TH) – the non-colligative depression of the freezing point, which does not affect the equilibrium melting point; (ii) dynamic ice shaping (DIS) where the shape of ice crystals is altered due to specific inhibition of difference faces on the ice surface; (iii) ice recrystallization inhibition (IRI) whereby the AF(G)Ps slow the rate of ice crystal growth (Ostwald ripening). The underlying mechanism of action for each individual property is still under investigation.6–8 Nishimura and co-workers synthesised several derivatives of AF(G)Ps, varying the carbohydrate and peptide components enabling them to identify the core motifs that were essential to maintain the TH and DIS activity.9 Combined with previous studies, which have also shown that AF(G)Ps are not tolerant to many structural modifications,1 it would seem that very particular requirements in terms of functional groups and tertiary structure are essential for AF(G)P activity. However, in most of these studies only TH and DIS were tested for, not IRI meaning its link to structure is less clear. In 2003 Enaide et al. observed that dramatically simplified glycoproteins could retain IRI activity but display essentially zero TH or DIS.10 This observation suggested that there might be multiple molecular mechanisms that give rise to the same macroscopic properties and that it is possible to design new molecules that specifically display IRI activity.11 Several glycopeptides and even small molecules have since been identified with varying degrees of IRI activity.Analysis of the process of cellular cryopreservation has revealed that ice growth during thawing is a major contributor to cell death.12 Consequently, any new materials that can inhibit ice growth (i.e. IRI activity) may find application to enhance the storage of urgently needed donor tissue for transplantation. In 2013 in the USA, 118000 individuals were on organ waiting lists and demand almost always outstrips supply.13,14 New regenerative medicines, or drug screening methods, based on stem cells also require improved (cryopreservation) storage methods.15,16 Attempts at cryopreservation with AF(G)Ps have reported varying degrees of success. Addition of AF(G)P to erythrocytes gave some cryopreservation enhancement, but above a critical concentration, its benefits stopped, limiting it use.17 This was found to be due to the formation of needle-like (spicular) ice crystals due to the DIS/TH activity of AF(G)Ps. Subsequent studies on the addition of AF(G)P to sperm resulted led to decreased motility18 and there is evidence that AF(G)P may be toxic to human cells.19 There are several other conflicting studies showing both benefits and problems of AF(G)Ps in cryopreservation, normally due to ice shaping, which have prevented their application.20,21 IRI-specific synthetic glycopeptides showed a marginal benefit in the cryopreservation of human embryonic liver cells.22
Due to the difficulties in synthesising glycopeptides,23 AF(G)P mimics are required, especially if they specifically reproduce only IRI for cyropreservation. Inada et al.24 and Gibson et al.25,26 have shown that poly(vinyl alcohol), PVA, is an extremely potent IRI despite having little or no structural similarity to AF(G)P. PVA with as few as 20 repeat units (MN = 880 g mol−1) can inhibit ice growth at concentrations below 1 mg mL−1. Gibson and co-workers showed that addition of just 1 mg mL−1 PVA to erythrocytes enabled their cryopreservation without the need for any organic solvents,27 such as glycerol or DMSO, which are themselves cytotoxic at high concentrations and not suitable for transfusion.
Synthetic polymers are particularly appealing as biomimetics due to their scalable synthesis, tuneable structure, and huge monomer scope, and have been employed as mimics of mussel adhesive proteins,28 antibacterial peptides29 and glycoproteins,30,31 for example. Despite this appeal, very few synthetic polymers have been demonstrated (quantitatively) to have IRI activity and it has become clear that simply having a poly(hydroxylated) structure (such as PVA) is not sufficient to guarantee IRI.32 Hydrophobicity is known to be a crucial component, with crystal structures of native AFPs showing defined hydrophobic and hydrophilic domains.2,33 Chemical modification of PVA (hydrophobic/hydrophilic substituents) also removes activity meaning that solution or surface absorbed conformation maybe important and there are few other lead structures to design new IRI-active compounds. Most polymers tested (in our hands) simply have no activity. Intriguingly, Matsumura et al. observed that poly(ampholytes) (which have both positive and negative charges) are very effective cryoprotectants, enabling solvent-free cryopreservation of stem cells.34 They observed that the 50% carboxylated poly(lysine) had very weak ice shaping effects (DIS). Similar results have been observed for ampholytic polysaccharides and vinyl polymers, but their IRI activity has not been quantitatively studied.35,36
The aim of this study was therefore to investigate the poly(ampholyte) structure as a new motif for IRI active polymers which would open the door to a diverse range of non-PVA based AF(G)P mimetic polymers with highly tuneable structures and properties. Such biomaterials will help address the urgent needs of tissue storage, transport and logistics,13 and lead to real clinical benefits building on our recent observations of the link between IRI and cell survival in cryopreservation.
Results and discussion
In order to obtain structure–property relationships between mixed charge polymers (polyampholytes) and their ice recrystallization inhibition activity it was necessary to devise a synthetic strategy that was compatible with controlled (radical) polymerization. The planned synthetic strategy is shown in Scheme 1, using poly(aminoethyl methacrylate) (PAEMA) derived from RAFT polymerization as the cationic polymer, as an alternative to poly(lysine). Reaction with succinic anhydride can then produce the mixed charged polymer, inspired by carboxylated poly(lysine), a known cryoprotectant.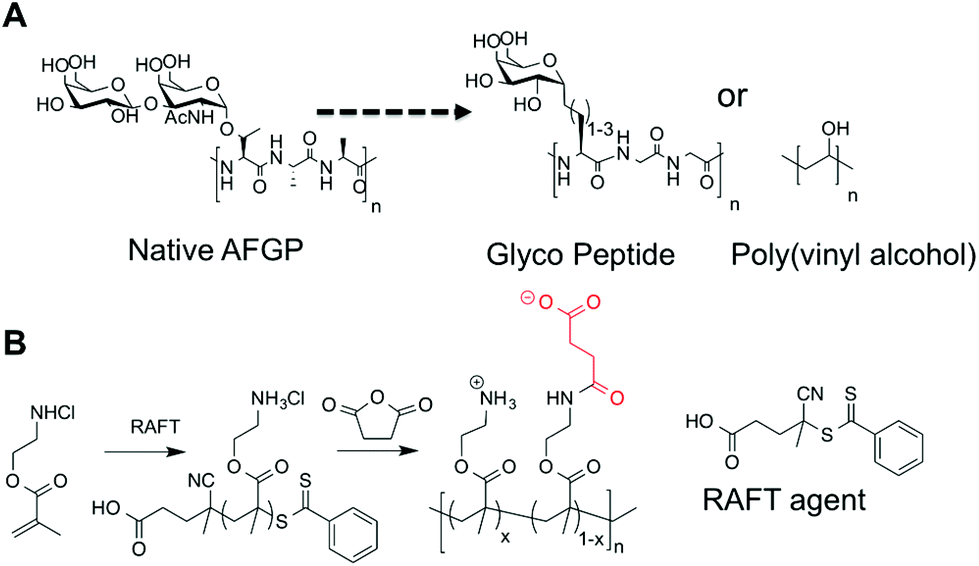 | ||
| Scheme 1 (A) Native AFGP and previously reported IRI active macromolecules; (B) synthetic method used in this report to obtain poly(ampholytes). | ||
A library of PAEMAs were synthesized using 4-cyanopentanoic acid dithiobenzoate (CTP) as the chain transfer agent, with 4,4′-azobis(4-cyanovaleric acid) (ACVA) as the radical source. A constant [monomer]:[CTA] ratio was employed in all experiments, with the polymerization time being varied to provide control over chain length. The monomer conversion was determined by 1H NMR spectroscopy against an internal mesitylene standard. All polymers were characterized by 1H NMR to provide an estimation of MN, by comparing the benzylic resonances derived from the RAFT agent to the polymer backbone, Table 1. Size exclusion chromatography (SEC) of these polymers was attempted in a range of aqueous buffers, but in all cases very broad dispersities were obtained, which can be ascribed to the well known self-reactivity of PAEMA which can lead to cross-linking37 (ESI†). To reduce this polymers were not handled in aqueous solution unless essential, and the solutions were acidified to ensure protonation of the amine groups. Previous reports of the polymerization of PAEMA suggest that it is a controlled process under identical RAFT conditions,38 and the values for MN extracted by end-group analysis are in agreement with that from conversion, suggesting the RAFT mediating agent is present at the chain ends. From this point on, the molecular weight is defined as that obtained from end group analysis. However, there must be assumed to be error in these measurements which are considered when interpreting data (below).
| Entry | Time (hours) | Conversiona (%) | M N,Theo (g mol−1) | M N,NMR (g mol−1) |
|---|---|---|---|---|
| Ratio [Monomer]:[CTA]:[I] = 400:1:0.2 was used in all polymerizations.a Determined by 1H NMR spectroscopy.b Theoretical MN determined from monomer feed ratio and conversion (1H NMR).c Mn determined by end group analysis. |
||||
| 11k | 0.75 | 8.2 | 5400 | 11000 |
| 20k | 1 | 18 | 11900 |
20000 |
| 23k | 2 | 35 | 23100 |
23000 |
| 27k | 3 | 44 | 29100 |
27000 |
| 33k | 3.5 | 52 | 34400 |
33000 |
| 40k | 5 | 61 | 40400 |
40000 |
| 67k | 6 | 74 | 49000 |
67000 |
In order to generate mixed-charge polymers with pendant carboxylic acid groups ring-opening of succinic anhydride was employed, Scheme 1. All the polymers in Table 1 were functionalized in this manner to give approximately 50 mol% (±5%) COOH functionality (ESI†). In order to screen the effect of COOH density on IRI activity (vide infra) 33 kg mol−1 PAEMA was functionalized at a range of COOH densities, Table 2. The degree of functionalization in each case was estimated by 1H NMR spectroscopy. 50 mol% carboxylated poly(lysine) was also synthesized from commercial poly(lysine) by the same method as a positive control.
| Entry | M N, PAEMA (kg mol−1) | Succinic anhydride (mol%) | f COOH (mol%) | M N copolymer (kg mol−1) |
|---|---|---|---|---|
| a Determined from PAEMA (CH2–CH2) to succinic anhydride ratio. | ||||
| P1 | 11 | 80 | 50 | 15.2 |
| P2 | 20 | 80 | 50 | 27.6 |
| P3 | 27 | 80 | 50 | 37.4 |
| P4 | 33 | 80 | 50 | 45.7 |
| P5 | 44 | 80 | 50 | 60.9 |
| P6 | 67 | 80 | 50 | 92.7 |
| P7 | 33 | 0 | 0 | 33.0 |
| P8 | 33 | 20 | 18 | 37.5 |
| P9 | 33 | 50 | 32 | 41.1 |
| P10 | 33 | 70 | 40 | 43.1 |
| P11 | 33 | 80 | 50 | 45.7 |
| P12 | 33 | 90 | 56 | 47.2 |
| P13 | 33 | 100 | 60 | 48.2 |
| P14 | 33 | 125 | 63 | 49.0 |
| P15 | 33 | 150 | 76 | 52.3 |
| P16 | 33 | 175 | 82 | 53.8 |
With this large panel of polymers at hand, quantitative ‘splat test’ measurements were undertaken to correlate polymer structure to ice recrystallization inhibition activity. Briefly, this assay involves dropping a small volume (∼10 μL) of phosphate buffered saline containing the polymers onto a CO2(S) cooled glass slide sat on an aluminium plate. This generates large numbers of very small (<10 μm) ice crystals which are allowed to grow at −6 °C for 30 minutes before being photographed through crossed polarizers. The mean largest grain size (MLGS) is measured and reported relative to a PBS standard, with smaller ice crystal sizes indicating a more active compound. Poly(ethyleneglycol) (PEG) is used as a negative control polymer as it is well known to have no significant IRI activity.25,26 The first polymer to be tested was 50 mol% carboxylated poly(lysine), which has been shown to be a useful cryoprotectant but has never been quantitatively assessed for IRI activity, Fig. 2. This polymer was shown to only have very weak IRI activity, giving ∼60% MLGS at 20 mg mL−1 compared to PEG which gave ∼75% MLGS at the same concentration. This is significantly weaker than the most well known polymeric IRI compound, poly(vinyl alcohol) which can inhibit ice growth below 1 mg mL−1. However, this still represents one of only a handful of polymers with definitive IRI activity and is an interesting lead structure for further investigation especially considering the known cryoprotectant properties of carboxylated poly(lysine) and their significant structure differences to AF(G)Ps. Encouraged by these results, a relatively long PAEMA (P6) 67 kg mol−1 with 50 mol% COOH was assessed for activity at a variety of concentrations. Pleasingly, this polymer showed significant IRI activity at concentrations up to 20 mg mL−1, with an MLGS of ∼40%, significantly more active than the corresponding poly(lysine) derivative. Example micrographs of ice wafers grown with P4 are shown in Fig. 1, and graphically in Fig. 2.
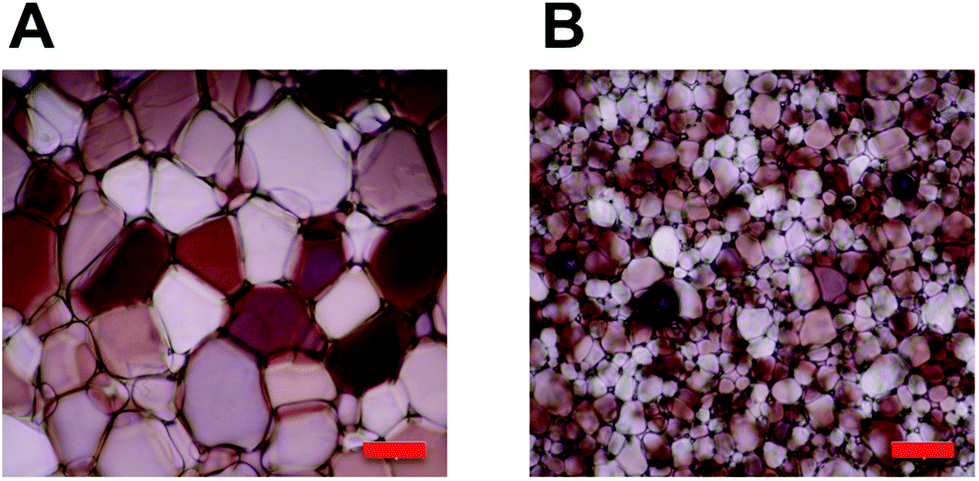 | ||
| Fig. 1 Cryomicrographs of ice wafers which have been annealed at −6 °C for 30 minutes. (A) PBS control; (B) P4, 20 mg mL−1. Scale bar = 100 μm. | ||
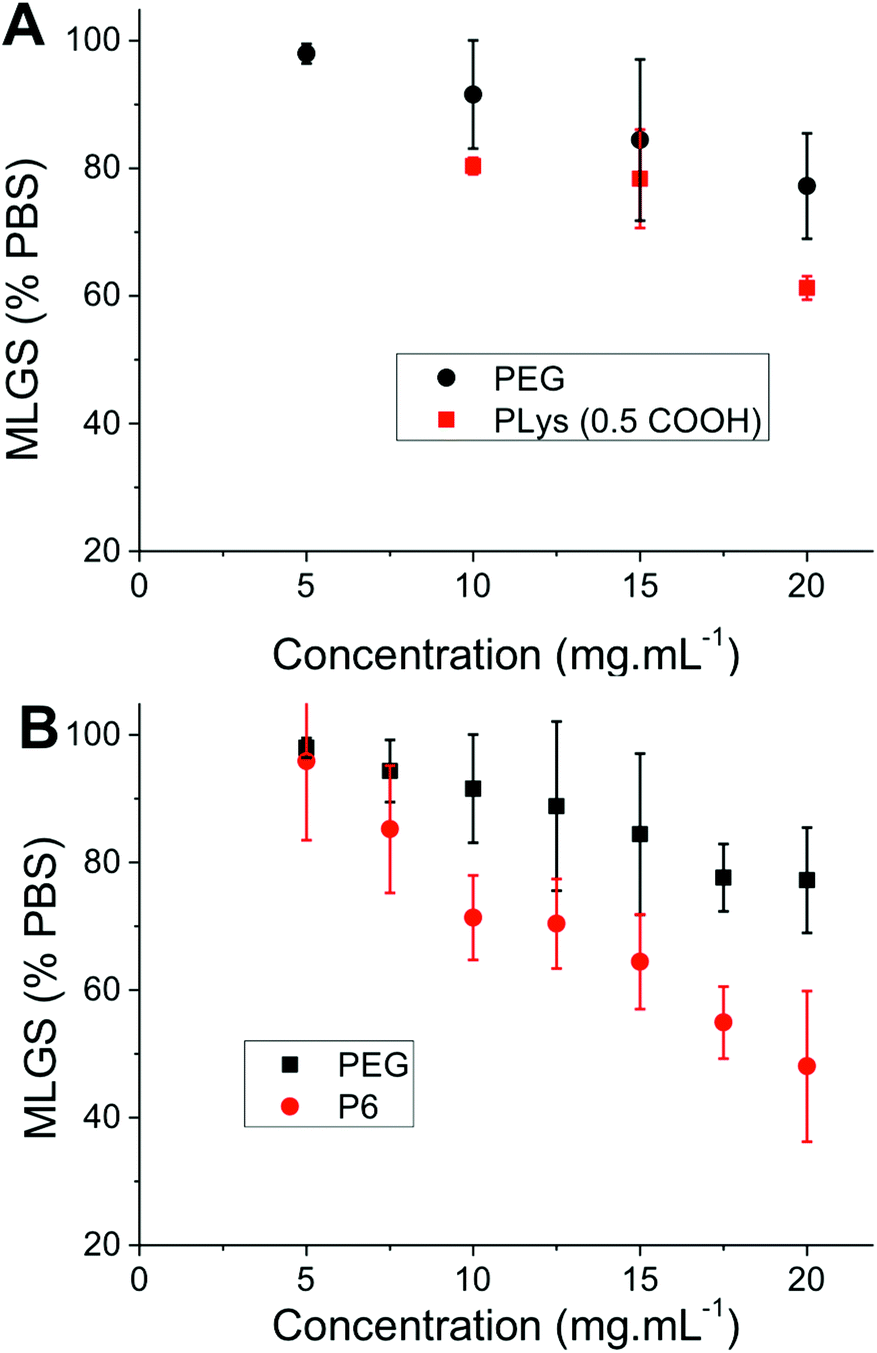 | ||
| Fig. 2 Ice recrystallization inhibition by poly(ampholytes). (A) Comparison of PEG and carboxylated poly(lysine); (B) comparison of PEG to PAEMA-co-succinic anhydride (50% functionality), P6, Error bars represent ± SD from a minimum of 3 repeats. MLGS = mean largest grain size relative to phosphate buffered saline control. | ||
To ensure that the observed IRI activity was due to the mixed-charge nature of the polymers, P7–P16 with varying degrees of carboxylation were investigated, Fig. 3. Polymers with 0 to 90% COOH (the highest achievable using this synthetic method) showed no IRI activity. As the density of COOH groups approached 50 mol% the IRI activity steadily increased (smaller crystals) demonstrating that the ratio of the two side chains is the crucial structural motif for activity. This is rather remarkable, and questions the mechanism of action of IRI-active polymers which are assumed to need defined ice-binding motifs (hydroxyls) which are not present here. Almost all other IRI active materials tested (PVA, native AFGP, carbohydrates) show a strong molecular weight dependence on activity, with larger polymers having more IRI. Fig. 3B shows the molecular weight dependency on IRI for the 50% COOH polymers, P1–P6. The shortest polymer used here (10 kg mol−1) had essentially zero IRI, and activity steadily increased as molecular weight increased, with the longest polymer (67 kg mol−1) giving average ice grain sizes half that of the shortest (10 kg mol−1) (which is equal to 4 fold decrease in ice area).
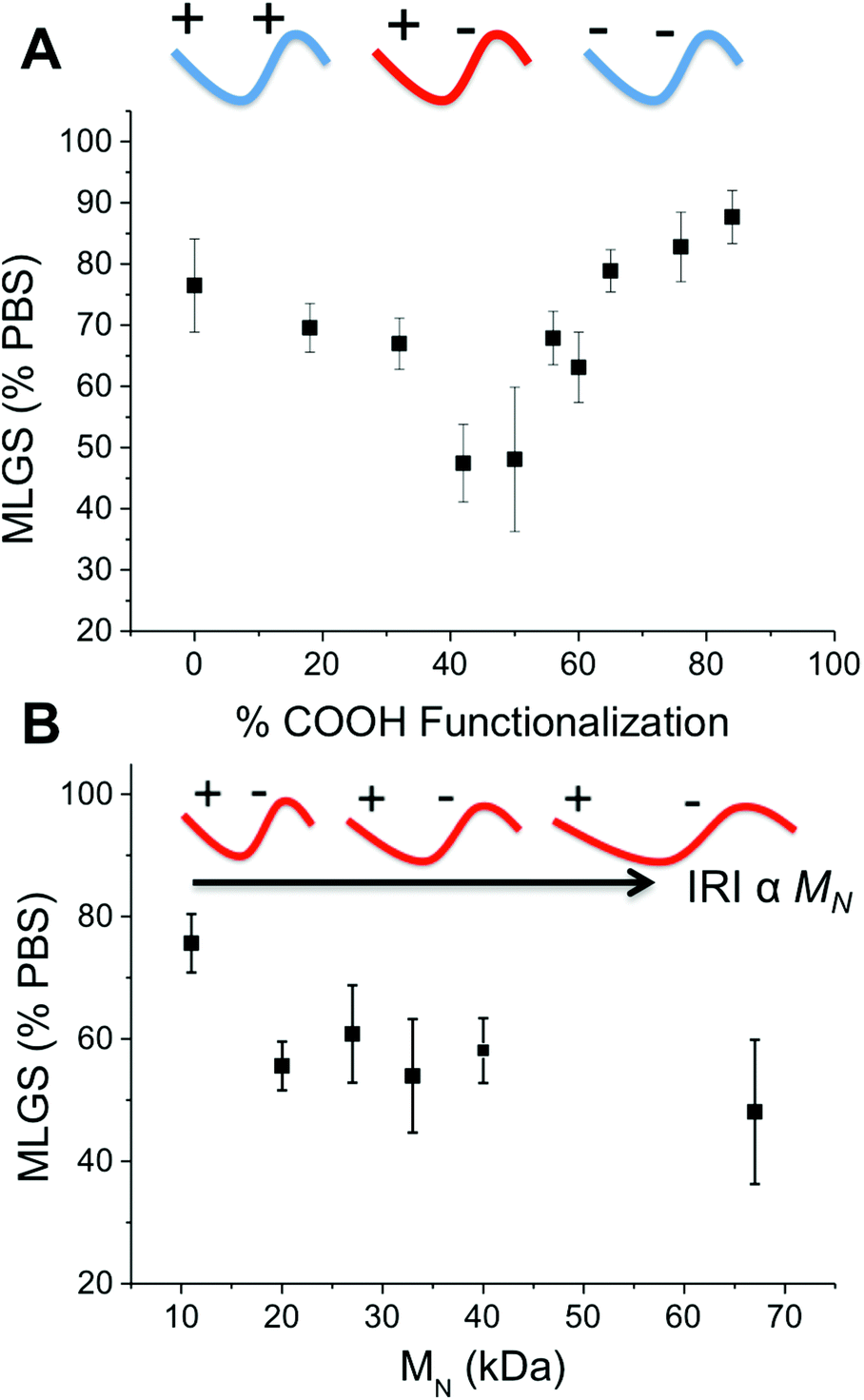 | ||
| Fig. 3 Ice recrystallization inhibition activity of PAEMA-derived polymers measured at 20 mg mL−1; (A) effect of degree of carboxylation on activity; (B) effect of molecular weight. Error bars represent ± SD from at least 3 repeats. MLGS = mean largest grain size relative to phosphate buffered saline control. | ||
There are currently no satisfactory models for explaining the mechanism by which IRI active polymers slow ice crystal growth, which has limited their a priori design. To test if this mixed-charge motif is a universal method for obtaining IRI polymers, a series of other polymers bearing multiply charged species were investigated. Poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) and poly([2-(methacryloyloxy) ethyl]dimethyl-(3-sulfopropyl) ammonium hydroxide) (PMEDS) were synthesized by RAFT polymerization of their corresponding monomers, Fig. 4A. PMPC was obtained with MN = 25 kg mol−1 and MW/MN = 1.12, and PMEDS MN = 21 kg mol−1 and MW/MN = 1.10. These monomers contain cationic and anionic charges on every repeat unit, but of a different nature to the PAEMA derivatives. Quantitative IRI testing revealed that both of these poly(betaines) had essentially zero IRI activity, with their observed MLGS being statistically identical to the negative PEG control at concentrations up to 15 mg mL−1. At 20 mg mL−1 pMPC had a weak enhancement which was only statistically different from PEG at a 95% confidence level. This implies that the distribution and nature (COOH, SO3) of the charges is crucially important, and will be the subject of future investigations.
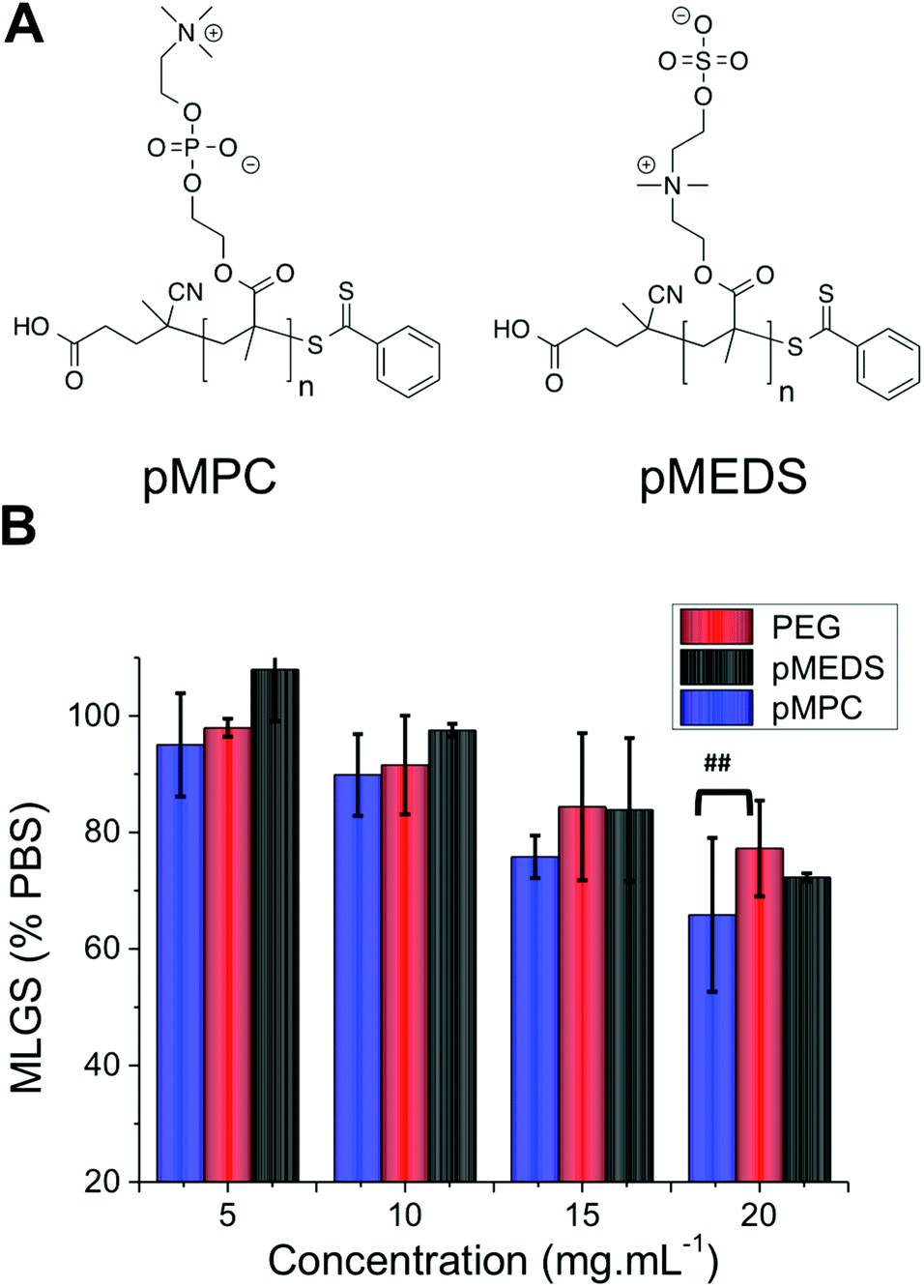 | ||
| Fig. 4 (A) Structures of pMPC and pMEDS; (B) ice recrystallization inhibition activity. Error bars represent ± SD from a minimum of 3 repeats. MLGS = mean largest grain size relative to phosphate buffered saline control. ## indicates a statistical difference (p < 0.05) according to students T-test. | ||
As a final test to determine if the mixed side-chain approach is a universal one, a series of carbohydrate-centred polymers with mixed charges were synthesised (ESI†).39 Briefly, a series mono, di and tri-saccharides were reacted with methacrylic anhydride to install a reactive methacrylate group on each of the hydroxyls. Addition of L-cysteine to the methacrylated cores by Michael addition (‘thiol–ene click’) enabled introduction of both an amine and carboxylic acid group at each position ensuring a discrete number of functional groups which is not possible using radical polymerizations (Table 3).
| Core sugar | Valency | M N,Theo (g mol−1) |
|---|---|---|
| a Assuming complete functionalization with cysteine. | ||
| Glucose | 5 | 1126 |
| Cellobiose | 8 | 1855 |
| Stachyose | 15 | 2690 |
The two smallest polymers (with either 5 or 8 side chains) displayed very little IRI activity that was statistically identical to PEG. However, the longest oligomer with 12 side chain units gave statistically significant smaller MLGS values compared to PEG and the other carbohydrate-based polymers. Whilst the actual values obtained indicate that these polymers have relatively low activity, it allowed the precise role of valency to be probed, showing that longer polymers are essential when using mixed-charge IRI polymers, but that the concept is a universal one. The structure of these polymers is also desirable as they are composed simply of carbohydrates and amino acids, which is appealing for biomedical applications (Fig. 5).
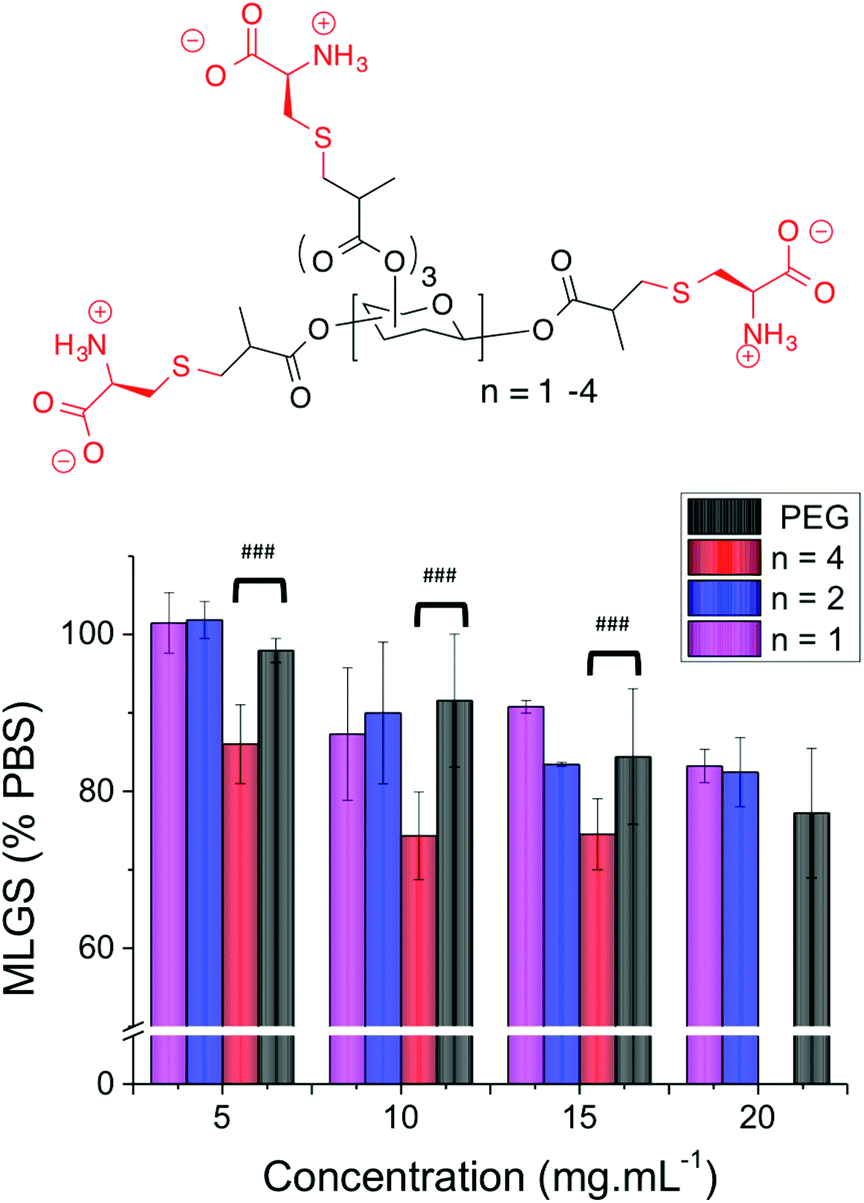 | ||
| Fig. 5 (A) Structure of carbohydrate-centred poly(ampholytes); (B) ice recrystallization inhibition activity. Error bars represent ± SD from a minimum of 3 repeats. MLGS = mean largest grain size relative to phosphate buffered saline control. ### indicates a statistical difference (p < 0.01) according to students T-test. | ||
The data presented above clearly demonstrate that mixed charge side chain polymers can inhibit ice growth, but still does not answers the many questions which remain regarding their mode of action. Gibson and Ben have both shown that hydrophobicity, without aggregation, is crucial in the design of new IRIs.11,32,40 This questions the commonly held theory that hydroxyl groups which are regularly spaced to interact directly with the ice crystal lattice are the crucial motifs to get IRI (or indeed thermal hysteresis and dynamic ice shaping).41 Terahertz spectroscopy investigations suggest that AF(G)Ps can disrupt the quasi-liquid layer (interfacial water) prevent exchange of water molecules and hence inhibiting growth.6,8 This mechanism would seem to require a hydrophobic domain to either disrupt the water structure or to bind to hydrophobic faces on the ice surface. AFP binding to ice has recently been speculated to be due to anchored clathrate-like water molecules using both hydrophobic interactions well as crystal-matching.42 A method to probe hydrophobic domains in polymers is dye inclusion assays. Addition of a hydrophobic dye that does not fluoresce in aqueous solution, but does in non-polar environments (such as pyrene) allows the ability of a polymer to solubilize hydrophobic compounds to be measured. This is the basis of critical micelle determination for self-assembling amphiphiles, which require a hydrophobic domain. The (polymer) concentration dependant fluorescence of several of the polymers described here were measured and compared to PVA, Fig. 6. Addition of PVA at less than 10 mg mL−1 lead to significant fluorescence, highlighting its ability to present a hydrophobic domain (and hence its application in colloidal stabilisers43). Quantifying the amount of dye incorporation is challenging due to potential self-quenching effects and non-linear fluorescence response, but we would estimate that the PVA can incorporate up to half the dye applied (43 μM) at the highest concentration tested. The non-IRI active PMPC gave rise to fluorescence of ∼2500 units at concentrations up to 50 m mL−1, significantly lower than observed for PVA. P3 showed essentially no fluorescence enhancement relative to PMPC, suggesting that P3 does not present a hydrophobic face in solution to solubilise the dye. Dynamic light scattering did not indicate micellisation in any cases. This testing does not rule out specific interactions with an ice surface though, nor does it suggest that additional hydrophobicity would not enhance activity, but rather than mechanistically the ampholyte structure does not induce a hydrophobic face. Indeed, studies by Matsumura et al. suggest that adding hydrophobicity into poly(ampholytes) can enhance their cryoprotective effect, but the influence of this on IRI has not been determined.36 The charge-balanced nature of the poly(ampholytes) may be able to disrupt the quasi-liquid layer (or pre-melt layer) at the ice–water interface which is hypothesised to inhibit ice growth, but direct measurements of this challenging interface are non-trivial. Future work will focus on unravelling the structural requirements of IRI activity, and delineating the observable macroscopic properties from the underlying molecular-level mechanisms, as well as probing their application in cryopreservation.
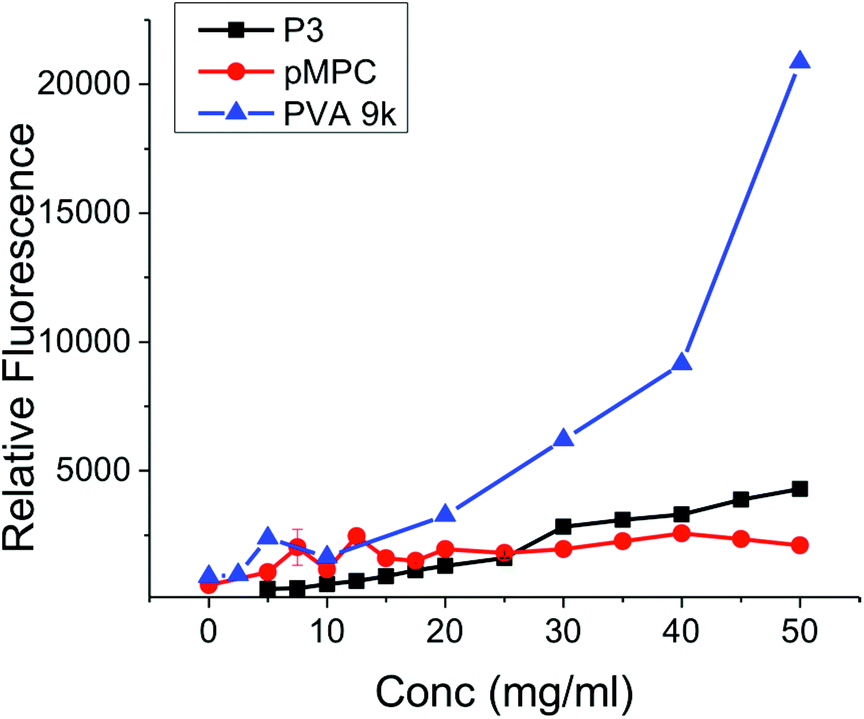 | ||
| Fig. 6 Dye inclusion assay using diphenyl hexatriene. λex = 360 nm λemission at 460 nm. | ||
Conclusions
This study represents the first detailed analysis of poly(ampholytes) as AF(G)P-mimetic, IRI active polymers. The significance of this work lies in the fact that the poly(ampholyte) structure is synthetically very accessible, but also very different to all previously recorded IRI-active polymers which are thus far limited to poly(hydroxylated) structures. It was demonstrated that the ratio of cationic to anionic groups was crucial, with a 1:1 balance giving maximal IRI activity, and that longer polymers had enhanced activity. The nature of the cation/anion was also found to be important as poly(betaines) showed no appreciable activity.
The remarkable activity of these polymers opens the door to the rational design of new, IRI-active polymers, which are not based on hydroxyl-group presentation and can be obtained from both vinyl and biological backbones. This also questions the underlying mode of action of IRI-active compounds, challenging the paradigm of precise-ice face recognition by hydroxyl groups. Ultimately, the development of IRI active polymers will impact regenerative medicine through enhanced cryopreservation of donor (stem) cells, or even whole organs.
Experimental section
Materials
Aminoethyl methacrylate (AEMA), 2-methacryloyloxyethyl phosphorylcholine (MPC), 4-cyanopentanoic acid dithiobenzoate (CTP), 4,4-Azobis(4-cyanovaleric acid) (V-501), succinic anhydride, ethylene sulphate, methacryloyloxy ethyl dimethyl-(3-sulfopropyl)ammonium hydroxide (MEDS) poly-ε-lysine hydrobromide (30–70 kDa), poly(ethylene glycol) (PEG, 100 kDa size), trehalose, sucrose, diphenylhexatriene (DPH), acetic acid and sodium acetate were purchased from Sigma Aldrich (UK). PEG was dialyzed over 24 hours with 5 water changes, while all other reagents were used as received unless otherwise stated. Phosphate-buffered saline (PBS) solution was prepared using preformulated tablets (Sigma-Aldrich) in 200 mL of Milli-Q water (>18.2 Ω mean resistivity) to give [NaCl] = 0.138 M, [KCl] = 0.0027 M, and pH 7.4.Physical and analytical methods
1H and 13C NMR spectra were recorded on Bruker DPX-300 and DPX-400 spectrometers using deuterated solvents obtained from Sigma-Aldrich. Chemical shifts in ppm (δ) are reported relative to residual tetramethylsilane (TMS). Fluorescence spectrometry was undertaken using a Synergy HT multi-mode microplate reader (BioTek UK, Bedfordshire, UK).Aqueous SEC was attempt using two different conditions. The first of these was performed on an Agilent 1260 Infinity Quaternary system utilizing 0.1 M HCL with 0.1% TFA at a flow rate of 1 mL min−1 while column and detector temperatures were maintained at 35 °C. The instrument was fitted with a PL aquagel guard column (50 × 7.5 mm, 8 μm) and a PSS NOVEMA cationic column (300 × 8.0 mm, 10 μm), The data was analyzed using Agilent GPC/SEC software. The second was performed on Cationic (acidic) aqueous GPC was performed on a Shimadzu Prominence UPLC system fitted with a differential refractive index detector. The eluent was 1 M acetic acid containing 0.3 M NaH2PO4 (pH 3) at a flow rate of 1 mL min−1 with column and detector cell temperatures maintained at 35 °C. The instrument was fitted with a Polymer Labs Aquagel-OH guard column (50 × 7.5 mm, 8 μm) followed by a pair of PL Aquagel-OH columns (30 and 40, 300 × 7.5 mm, 8 μm). Column calibration was achieved using narrow poly(2-vinyl pyridine) standards (Polymer Standards Service, Germany) of known molecular weight in the range 0.8–256 kDa. Molecular weights and dispersity values were calculated using Shimadzu LabSolutions software with GPC analysis add-on.
Synthesis of poly(amino ethyl methacrylate), PAEMA
Aminoethyl methacrylate monomer was polymerised by aqueous reversible addition–fragmentation chain transfer (RAFT) polymerisation as previously detailed.38 The monomer to CTA ratio was 400:1 while CTA to initiator ratio was 5:1. AEMA monomer (825 mg, 5.4 mmol) V-501 (0.7 mg, 0.0025 mmol) and CTP (3.5 mg, 0.013 mmol) were dissolved in 0.6 mL of acetate buffer at pH 5.2 (produced using 0.27 mol L−1 acetic acid and 0.73 mol L−1 sodium acetate) in a 50 mL round bottomed flask, from a stock solution and subsequently diluted to 5 mL with the addition of further acetate buffer. The flask was purged with nitrogen for 45 minutes and placed in an oil bath at 70 °C. The reaction was allowed to proceed to produce polymers of varying molecular weight. After the required amount of time (30–540 minutes) the reaction was quenched using liquid nitrogen. Dialysis using acetate buffer (24 hours, 5 water changes) and lyophilisation were then used to purify the product. 1H NMR (D2O): δ 4.21 (br, 2H, –OCH2); δ 3.31 (br, 2H, –NH2CH2); δ 1.95 (br, 2H, –CH2); δ 0.83–1.36 (br, 3H, –CH3).
Acknowledgements
Equipment used was supported by the Innovative Uses for Advanced Materials in the Modern World (AM2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). MIG is a Birmingham Science City Interdisciplinary Research Fellow funded by the Higher Education Funding Council for England (HEFCE). This work was supported by a Research Project Grant from the Royal Society. DEM acknowledges the EPSRC for a studentship from the MOAC Doctoral Training Centre. This work was supported by EPSRC grant number EP/H005625/1.Notes and references
- M. M. Harding, P. I. Anderberg and A. D. J. Haymet, Eur. J. Biochem., 2003, 270, 1381–1392 CrossRef CAS.
- M. M. Harding, L. G. Ward and A. D. J. Haymet, Eur. J. Biochem., 1999, 264, 653–665 CrossRef CAS.
- A. P. Esser-Kahn, V. Trang and M. B. Francis, J. Am. Chem. Soc., 2010, 132, 13264–13269 CrossRef CAS PubMed.
- M. I. Gibson, Polym. Chem., 2010, 1, 1141–1152 RSC.
- V. Bouvet and R. N. Ben, Cell Biochem. Biophys., 2003, 39, 133–144 CrossRef CAS.
- K. Meister, S. Ebbinghaus, Y. Xu, J. G. Duman, A. DeVries, M. Gruebele, D. M. Leitner and M. Havenith, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1617–1622 CrossRef CAS PubMed.
- K. A. Sharp, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7281–7282 CrossRef CAS PubMed.
- A. B. Siemer, K.-Y. Huang and A. E. McDermott, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17580–17585 CrossRef CAS PubMed.
- Y. Tachibana, G. L. Fletcher, N. Fujitani, S. Tsuda, K. Monde and S.-I. Nishimura, Angew. Chem., Int. Ed., 2004, 43, 856–862 CrossRef CAS PubMed.
- A. Eniade, M. Purushotham, R. N. Ben, J. B. Wang and K. Horwarth, Cell Biochem. Biophys., 2003, 38, 115–124 CrossRef CAS.
- C. J. Capicciotti, M. Leclere, F. A. Perras, D. L. Bryce, H. Paulin, J. Harden, Y. Liu and R. N. Ben, Chem. Sci., 2012, 3, 1408–1416 RSC.
- A. Fowler and M. Toner, Ann. N. Y. Acad. Sci., 2005, 1066, 119–135 CrossRef CAS PubMed.
- A. Opar, Nat. Med., 2008, 14, 225 CrossRef CAS PubMed.
- R. A. Wolfe, E. C. Roys and R. M. Merion, Am. J. Transplant., 2010, 10, 961–972 CrossRef CAS PubMed.
- M. Richards, C.-Y. Fong, S. Tan, W.-K. Chan and A. Bongso, Stem Cells, 2004, 22, 779–789 CrossRef PubMed.
- J. G. Baust, D. Gao and J. M. Baust, Organogenesis, 2009, 5, 90–96 CrossRef.
- J. F. Carpenter and T. N. Hansen, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 8953–8957 CrossRef CAS.
- C. Koshimoto and P. Mazur, Cryobiology, 2002, 45, 49–59 CrossRef CAS.
- S. Liu, W. Wang, E. von Moos, J. Jackman, G. Mealing, R. Monette and R. N. Ben, Biomacromolecules, 2007, 8, 1456–1462 CrossRef CAS PubMed.
- L. O'Neil, S. J. Paynter, B. J. Fuller, R. W. Shaw and A. L. DeVries, Cryobiology, 1998, 37, 59–66 CrossRef PubMed.
- T. Wang, Q. Zhu, X. Yang, J. R. Layne and A. L. DeVries, Cryobiology, 1994, 31, 185–192 CrossRef CAS PubMed.
- M. Leclere, B. K. Kwok, L. K. Wu, D. S. Allan and R. N. Ben, Bioconjugate Chem., 2011, 22, 1804–1810 CrossRef CAS PubMed.
- B. L. Wilkinson, R. S. Stone, C. J. Capicciotti, M. Thaysen-Andersen, J. M. Matthews, N. H. Packer, R. N. Ben and R. J. Payne, Angew. Chem., Int. Ed., 2012, 51, 3606–3610 CrossRef CAS PubMed.
- T. Inada and S.-S. Lu, Cryst. Growth Des., 2003, 3, 747–752 CAS.
- M. I. Gibson, C. A. Barker, S. G. Spain, L. Albertin and N. R. Cameron, Biomacromolecules, 2009, 10, 328–333 CrossRef CAS PubMed.
- T. Congdon, R. Notman and M. I. Gibson, Biomacromolecules, 2013, 14, 1578–1586 CrossRef CAS PubMed.
- R. C. Deller, M. Vatish, D. A. Mitchell and M. I. Gibson, Nat. Commun., 2014, 5 Search PubMed.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS PubMed.
- K. Lienkamp and G. N. Tew, Chem. – Eur. J., 2009, 15, 11784–11800 CrossRef CAS PubMed.
- S.-J. Richards, M. W. Jones, M. Hunaban, D. M. Haddleton and M. I. Gibson, Angew. Chem., Int. Ed., 2012, 51, 7812–7816 CrossRef CAS PubMed.
- M. W. Jones, L. Otten, S. J. Richards, R. Lowery, D. J. Phillips, D. M. Haddleton and M. I. Gibson, Chem. Sci., 2014, 5, 1611–1616 RSC.
- R. C. Deller, T. Congdon, M. A. Sahid, M. Morgan, M. Vatish, D. A. Mitchell, R. Notman and M. I. Gibson, Biomater. Sci., 2013, 1, 478–485 RSC.
- S. P. Greather, M. J. Kulper, S. M. Gagnes, V. K. Walker, Z. Jia, B. D. Sykes and P. L. Davies, Nature, 2000, 406, 325–328 CrossRef PubMed.
- K. Matsumura and S.-H. Hyon, Biomaterials, 2009, 30, 4842–4849 CrossRef CAS PubMed.
- M. Jain, R. Rajan, S.-H. Hyon and K. Matsumura, Biomater. Sci., 2014, 2, 308–317 RSC.
- R. Rajan, M. Jain and K. Matsumura, J. Biomater. Sci., Polym. Ed., 2013, 24, 1767–1780 CrossRef CAS PubMed.
- A. Emileh, E. Vasheghani-Farahani and M. Imani, Eur. Polym. J., 2007, 43, 1986–1995 CrossRef CAS PubMed.
- A. H. Alidedeoglu, A. W. York, C. L. McCormick and S. E. Morgan, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5405–5415 CrossRef CAS PubMed.
- T. Congdon, C. Wilmet, R. Williams, J. Polt, M. Lilliman and M. I. Gibson, Eur. Polym. J. DOI:10.1016/j.eurpolymj.2014.06.001.
- A. K. Balcerzak, M. Febbraro and R. N. Ben, RSC Adv., 2013, 3, 3232–3236 RSC.
- C. Budke and T. Koop, ChemPhysChem, 2006, 7, 2601–2606 CrossRef CAS PubMed.
- C. P. Garnham, R. L. Campbell and P. L. Davies, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7363–7367 CrossRef CAS PubMed.
- A. Lee, H. Y. Tsai and M. Z. Yates, Langmuir, 2010, 26, 18055–18060 CrossRef CAS PubMed.
- C. A. Knight, J. Hallett and A. DeVries, Cryobiology, 1988, 25, 55–60 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- K. Matsumura and S.-H. Hyon, Biomaterials, 2009, 30, 4842–4849 CrossRef CAS PubMed.
- B. Yu, A. B. Lowe and K. Ishihara, Biomacromolecules, 2009, 10, 950–958 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: This includes full characterisation and synthetic details. See DOI: 10.1039/c4bm00153b |
| This journal is © The Royal Society of Chemistry 2014 |