
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biophysical regulation of hematopoietic stem cells
C.
Lee-Thedieck
*a and
J. P.
Spatz
bc
aKarlsruhe Institute of Technology (KIT), Institute of Functional Interfaces, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: cornelia.lee-thedieck@kit.edu; Fax: +49-721 608-23478; Tel: +49-721 608-23588
bMax Planck Institute for Intelligent Systems, Department of New Materials and Biosystems, Heisenbergstr. 3, 70569 Stuttgart, Germany
cUniversity of Heidelberg, Department of Biophysical Chemistry, Im Neuenheimer Feld 253, 69120 Heidelberg, Germany
First published on 17th July 2014
Abstract
Blood is renewed throughout the entire life. The stem cells of the blood, called hematopoietic stem cells (HSCs), are responsible for maintaining a supply of all types of fresh blood cells. In contrast to other stem cells, the clinical application of these cells is well established and HSC transplantation is an established life-saving therapy for patients suffering from haematological disorders. Despite their efficient functionality throughout life in vivo, controlling HSC behaviour in vitro (including their proliferation and differentiation) is still a major task that has not been resolved with standard cell culture systems. Targeted HSC multiplication in vitro could be beneficial for many patients, because HSC supply is limited. The biology of these cells and their natural microenvironment – their niche – remain a matter of ongoing research. In recent years, evidence has come to light that HSCs are susceptible to physical stimuli. This makes the regulation of HSCs by engineering physical parameters a promising approach for the targeted manipulation of these cells for clinical applications. Nevertheless, the biophysical regulation of these cells is still poorly understood. This review sheds light on the role of biophysical parameters in HSC biology and outlines which knowledge on biophysical regulation identified in other cell types could be applied to HSCs.
 C. Lee-Thedieck | Cornelia Lee-Thedieck is a junior group leader for Stem Cell–Material-Interactions at the Karlsruhe Institute of Technology (KIT), Institute of Functional Interfaces. From 2009 to 2012 she was a group leader and from 2007 to 2009 a postdoctoral fellow at the Max Planck Institute for Intelligent Systems in the department of Prof. Spatz. She received her Ph.D. in Biology in 2007 and her Diploma in Biochemistry in 2004 from the University of Tübingen. |
 J. P. Spatz | Joachim P. Spatz is the Director of the department of New Materials and Biosystems at the Max Planck Institute for Intelligent Systems and full Professor for Biophysical Chemistry at the University of Heidelberg. From 2000 to 2004 he was Associate Professor at the University of Heidelberg. In 2000 he received his habilitation in Physics at the University of Ulm. From 1997 to 1998 he was a postdoctoral fellow at the Institut Curie, Paris. He received his Ph.D. in Physics (1996) and his Diploma in Physics (1994) from the University of Ulm. |
Introduction
Stem cells have, by definition, the abilities to differentiate into specialized cell types and to self-renew. Among the adult stem cells (i.e. stem cells that are organ-specific, possess important roles in tissue maintenance and repair, and are present throughout the entire life) hematopoietic stem cells (HSCs) are the best-studied.1 HSCs, the stem cells of the blood system, renew our blood with billions of fresh cells such as erythrocytes, T- and B-cells every day.2 Since the 1960s, HSCs have been used in the clinical setting to treat patients that suffer from haematological disorders such as leukaemia or lymphoma.3 However, the supply of HSCs for clinical applications is limited in terms of the availability of matching donors (= number of available transplants) and the number of HSCs per transplant. The latter is the case in particular when umbilical cord blood is used as an alternative source of transplantable HSCs.4 Steps toward efficient HSC proliferation in vitro could severely contribute to an improved supply with HSCs for transplantation and thereby help tens of thousands of patients. However, this remains a challenge, as conventional cell culture systems thus far have failed to reproduce the efficiency of the natural HSC environment in terms of controlling cellular behaviour.In 1978, Schofield proposed the niche hypothesis for HSCs.5 Niches are highly specialized microenvironments in which the stem cells receive all the signals they need in order to maintain their stem cell character (i.e., the ability to self-renew and to differentiate). In the case of HSCs these niches are located in the red bone marrow of trabecular bones. Up until now, several locations and entities have been described as HSC niches and new factors, parameters and cell-types that play a role in HSC niches are continuously being discovered. This continuously adds complexity to the system.6 Despite the fact that for other stem cells, such as mesenchymal stem cells, the importance of physical parameters in stem cell biology has been widely accepted, surprisingly little is known on the impact of (bio)physical parameters on HSCs. The aim of this review is to give an overview on the current understanding of the biophysical regulation of HSCs and to discuss the possibilities of transferring existing concepts of biophysical regulation mechanisms to HSCs.
The stem cell niche
Stem cell niches are specific three-dimensional microenvironments that regulate adult stem cell survival, maintenance, proliferation and differentiation.7 The signals through which the niche controls stem cell function are (i) direct contact and communication between stem cells and their adjacent supporting niche cells, (ii) interactions with the extracellular matrix, and (iii) stimulation by soluble components such as growth factors, hormones, cytokines or chemokines.Studies have located HSC niches either close to the endosteum (‘endosteal niche’),8–10 adjacent to bone marrow sinusoids11 or next to arterioles12 (‘perivascular niches’). Current literature suggests that these are functionally distinct microenvironments that host distinct types of HSCs.6,13 Each of these niches is created by multiple cell types that contribute to niche function and HSC regulation.14 In other words, there is not a singular niche cell that regulates HSCs alone. Instead, the niches orchestrate the functions of multiple niche cell types in order to fulfil their task (Table 1).
| Category | Components | References |
|---|---|---|
| Extracellular matrix | Collagens type I, II, III, IV, V, VI, X | 25–27 |
| Laminins | 19,28 | |
| Fibronectin | 25,27,29 | |
| Nidogen | 30 | |
| Tenascin-C | 27,30 | |
| Thrombospondin | 26,27 | |
| Vitronectin | 26 | |
| Decorin | 26 | |
| Fibulin | 31,32 | |
| Hyaluronic acid | 33,34 | |
| Osteopontin | 21,22,35 | |
| Cellular components | Endothelial cells | 11 |
| Mesenchymal stem/stromal cells | 36 | |
| Perivascular cells | 6 | |
| CAR (CXCL-12 abundant reticular) cells | 37,38 | |
| Non-myelinating Schwann cells | 39 | |
| Macrophages | 40–42 | |
| Sympathetic nerves | 43 | |
| Osteoblasts | 8–10 | |
| Osteoclasts | 44,45 | |
| Osteocytes | 46 | |
| Physical parameters | Three-dimensional architecture | 29,47–51 |
| Mechanical properties | 52–54 | |
| Nanostructural features | 47,52,55–57 | |
| Shear stress/fluid flow | 58–60 |
A range of different cell populations have been used to study HSCs. They differ in terms of species (mainly mouse or human) as well as markers that were used for their purification. As a result, different hematopoietic subpopulations as well as cell populations that are more or less enriched with HSC subtypes have been applied. Whereas suitable markers for the identification and purification of HSCs are available in mice, such markers are still missing for human HSCs and it appears that the murine and human hematopoietic system have remarkable differences.15 Nevertheless, the term ‘HSC’ is used throughout this review for readability reasons. In order to acknowledge the potential impact of species and subpopulations on the biophysical regulation of HSCs, footnotes provide information on the cell populations used in the respective study wherever necessary.
The space between cells is filled by the extracellular matrix (ECM), which provides structural stability as well as anchorage sites for cell adhesion and migration. Furthermore, the ECM regulates cell functions such as morphology and development.16 The ECM is secreted by cells into the extracellular space. It is a complex composition of various molecules that can be categorized into two groups: (i) structural proteins (including glycoproteins) that provide structural integrity for tissues, and (ii) proteoglycans and glycosaminoglycans, which provide the capacity to hold substantial amounts of water and to bind growth factors.17
The structural proteins of the bone marrow ECM (including collagens, laminins and fibronectin) provide anchorage sites that are recognized by cells through specific cell adhesion molecules. In this way, the ECM plays a crucial role in the retention of stem cells in their niches as well as their mobilization from the niche.16 Integrins – which directly link the ECM with the intracellular actin cytoskeleton – are an important family of cell adhesion receptors in this context. Furthermore, laminins and fibronectin act not only as structural elements that provide anchorage sites for cells. They also influence HSC†,‡ proliferation, differentiation, and the cell's ability to engraft.18–20 Osteopontin – another member of the structural proteins of the bone marrow ECM – was shown to negatively regulate both the HSC pool size in vivo in mice as well as the in vitro proliferation of human HSCs.†21,22
Proteoglycans consist of a protein core with covalently bound glycosaminoglycans. Their ability to bind and present growth factors to stem cells contributes to cell adhesion and migration as well as playing a role in regulating differentiation.23 Among the glycosaminoglycans, hyaluronic acid is an important component of the bone marrow ECM that impacts HSC behaviour via interaction with its receptor CD44.24Table 1 contains a comprehensive list of known bone marrow ECM components.
Many ECM components are distributed throughout the bone marrow unevenly. They are more or less enriched in certain compartments (including compartments described to be potential HSC niches). While fibronectin appears to be expressed ubiquitously in the bone marrow27 (endothelial61 and endosteal25 regions), osteopontin and collagen type I are restricted to endosteal regions.21,25 Most laminin isoforms that are detected in the bone marrow are found in association with blood vessels, sinusoids and endothelial cells – in other words in vascular regions.19 Variations in ECM composition may contribute to the different functions of the bone marrow regions (and potential niche microenvironments). These functions might be elicited not only by differences in the biological and/or (bio)chemical properties of the ECM molecules but also by differences in their biophysical properties. For example, the substrate stiffness – characterized by the Young's modulus E – varies broadly in the different potential niche microenvironments. They range from very soft in the marrow (E = 0.3 kPa)62 to intermediate in endothelia (0.5–2.0 kPa)63 and vessel walls (5–8 kPa)64,65 to relatively stiff in the osteoid matrix (35 kPa).66
In spite of the progress made so far, characterizing the composition of the niche is still a matter of ongoing research and engineering an artificial stem cell niche is considered to be one of the major challenges in hematopoietic research.26 In the attempt to design and recreate stem cell niches, physical niche properties are emerging as important HSC influencing parameters that must be taken into account.
Environmental sensing
Cells sense their environment through specific receptors that can recognize their matching counterpart molecule (in other words, their ligand) when it is nearby. In addition to recognizing the type of ligand that is bound by the receptor, cells can also “feel” other environmental and ligand cues, such as rigidity or elasticity. In other words, the cell can integrate both the chemical nature of its environment as well as physical cues generated by external and internal forces. In this way cells can sense the mechanical properties of the surrounding tissue, nanostructural features, and shear forces.67Fig. 1 gives a schematic overview of the different biological, chemical and physical stimuli in the environment of stem cells.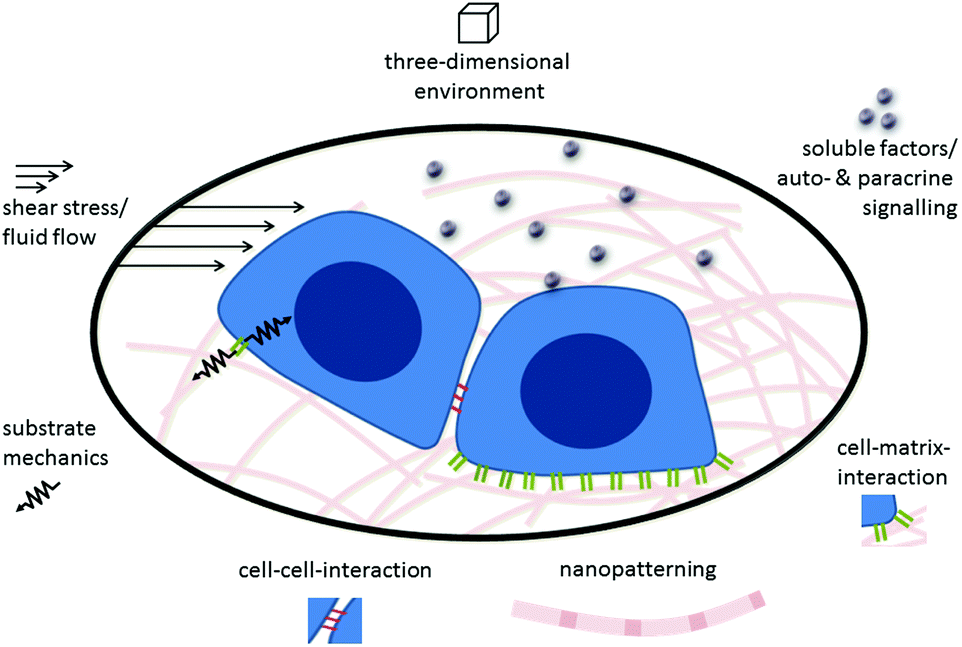 | ||
| Fig. 1 Environmental stimuli that influence cells. Cells are naturally localized in three-dimensional environments. In their in vivo environments they are subjected to biochemical and biological signals elicited by cell–cell interactions including direct contacts and communication through soluble factors as well as cell–matrix interactions. At the same time, cells are stimulated by the physical parameters of the environment, such as substrate mechanics, shear forces and the nanopatterning of the surrounding matrix. | ||
Cells in living tissues are closely connected to other cells and the ECM via cell adhesion molecules (CAMs). These molecules connect the cell's interior to its external environment and typically consist of an extracellular domain, a transmembrane region and an intracellular domain that elicits intracellular signalling or interacts with the cytoskeleton of the cell. Most CAMs can be grouped into distinct families: integrins, cadherins, selectins and the immunoglobulin superfamily.68 Integrins are the most common cellular receptors for ECM molecules. They are heterodimeric molecules, each composed of one alpha and one beta chain.69,70 The 18 different alpha and 8 beta chains identified so far dimerize non-covalently to form the 24 integrins known today.71 The extracellular domain of integrins recognizes and binds to specific ECM molecules. The intracellular domain is linked to the actin cytoskeleton and also associates with signalling molecules such as kinases and phosphatases.72,73
Signalling through integrins is a bi-directional process that functions both outside-in and inside-out.70 Upon binding of an ECM element to the extracellular part of the integrin receptor, the cell receives information on the biochemical and biophysical properties of its environment. Integrins, in their function as signal transmitters, then trigger an intracellular cascade of signalling events, including phosphorylation and signalling through small G-proteins (outside-in signalling). Local cytoskeleton dynamics are changed in response, which in turn leads to the generation of force. These processes lead to directly observable alterations, e.g., in cell shape and motility, and later cause changes on the transcriptional level. This ultimately affects cell proliferation, differentiation and survival.67 The control over the affinity of integrins to their extracellular ligands (as part of their adhesion molecule function) also occurs via signals which originate from the cell's interior and act on the cytoplasmic tail of the integrins. These signals trigger conformational changes in the extracellular ligand-binding domain of the receptor and thereby lead to changes in ligand affinity (inside-out signalling).74
When integrins establish contact with the ECM nascent adhesive structures, so-called focal complexes,75 appear. In anchorage-dependent cells such as fibroblasts these focal complexes are transient. They can disappear with time or mature into larger, more stable adhesive structures – the focal adhesion. Mature focal adhesions have an elongated appearance and are found at the ends of actin stress fibres.67,76 It appears that integrin-mediated contacts to the ECM formed by HSCs differ from those found in obligatory adherent cells, because the actin cytoskeleton of HSCs does not develop stress fibres.77 Although HSCs† express zyxin78 – a marker that is considered specific for mature focal adhesions and that is not detected in nascent focal complexes79 – it still seems most likely that HSCs do not form mature focal adhesions. Nevertheless, many of the substrates that were shown to elicit mechanosensitive responses in HSCs do actually target integrins.52,54,62 This suggests that, similar to their role in anchorage-dependent cells, integrins are likely to act as environmental sensors in HSCs, even though the composition and maturation of the associated multiprotein complexes might differ.
These differences are accompanied by some striking phenomenological differences between HSCs and anchorage-dependent cells. HSCs,† instead of showing spreading behaviour on 2D surfaces, polarize and form a uropod.80–82 They exhibit an amoeboid-type migration behaviour, which leads to a fast migration speed of several μm per minute.54 These phenomenological observations in 2D cell culture systems indicate a more transient nature of integrin-mediated contacts between ECM molecules and HSCs, compared to contacts with anchorage-dependent cells.
Integrin–actin link
The formation of adhesive structures that link cells to the ECM first requires integrin activation. This involves a conformational re-organization of the dimer, resulting in an enhanced affinity to its ligand.83 The connection of the cytoplasmic domain of the integrin beta-chain with the actin cytoskeleton via kindlins and talins is a crucial step during this activation process.84–88 As described for anchorage-dependent cells, it has also been shown for HSCs§ that adhesion to the ECM is talin-dependent.89 HSCs are assumed to express kindlin-3 (synonym: FERMT3), because this member of the kindlin family is specifically found in hematopoietic cells and platelets, including hematopoietic organs during murine fetal development and bone marrow mononuclear cells in adult mice.90,91Following the engagement of talin and kindlin, vinculin binds to talin and fosters the clustering of multiple activated integrins. Vinculin also strengthens the integrin–actin cytoskeleton linkage by binding to actin through its tail domain.92,93 However, experiments in which vinculin expression was silenced have provided evidence that vinculin is actually not necessary for integrin-dependent adhesion in HSCs,§ but is indispensable for integrin-independent repopulation of the bone marrow by HSCs.§89 These results indicate that vinculin plays a different role in HSCs than in anchorage-dependent cells.
Actin assembly and flow lead to cell locomotion. In non-muscle cells actin movement depends on non-muscle myosin II activity. Non-muscle myosin II exists in two isoforms (IIA and IIB) both of which are expressed by HSCs. Both isoforms show a cortical localization in freshly isolated, uncultured HSCs.† Myosin IIB accumulates in the cortical regions of the uropod during the process of adhesion-induced polarization, whereas myosin IIA localization is diffuse in the cytoplasma. Isoform switching seems to regulate HSC differentiation and motility.62,77
Motility and migration are essential to HSC function in embryogenesis and adulthood.94 However, the cytoskeletal motility apparatus of HSCs has not been investigated in great detail, as of yet. Gene expression analysis of HSCs† using cDNA arrays, e.g., revealed the expression of several members of this apparatus, namely alpha-actinin, dynein, dynamin 2, dynactin 1 and drebrin 1.95 Among these, alpha-actinin is of particular interest in the context of biophysical regulation of cells, because it is involved in the integrin-mediated force transmission.96 Its role in adhesion and force transduction in HSCs is yet to be elucidated.
Integrin-mediated signalling
Besides connecting the receptor to the actin cytoskeleton, the intracellular multiprotein complex around ligand-bound integrin clusters is also involved in transmitting signals into the cell. Many different components of the integrin ‘adhesome’ have been described and their interplay is highly complex.97 Here, we will focus on some key elements that are known to play a role in HSCs and/or the mechanotransduction of stem cells.The most prominent integrin-linked signalling molecule is the focal adhesion kinase (FAK), which is a non-receptor tyrosine kinase. It is a key element of many signalling pathways that lie downstream of integrins. FAK plays a central role in adhesion turnover, the activation of small GTPases of the Rho family, and crosstalk between integrin- and growth factor receptor mediated signalling.98 FAK interacts with talin and paxillin.99–101 It binds and is involved in the phosphorylation and activation of its substrates including paxillin and guanine nucleotide exchange factors (which are important for signalling via small G-proteins).102 The FAK homologue Pyk2 (synonym RAFTK) has been described as the principal ‘focal adhesion kinase’ in HSCs.†103 Pyk2 shares many properties with FAK. However, these kinases are not redundant and do possess individual capacities.69 Both, Pyk2 and FAK are necessary for proper HSC function.
Subsequent to integrin–ECM interaction, a further family of non-receptor kinases – the Src family kinases (SFK) – is quickly activated. These can either bind to the cytoplasmic tail of the integrin β-chain directly or to FAK. They can phosphorylate FAK and FAK-associated proteins.69 The SFKs Lyn, Hck and Fgr are specific to the hematopoietic compartment and play a role in cytokine-induced mobilization of HSCs§ from the bone marrow to the circulation as well as in the engraftment of transplanted HSCs¶ to the bone marrow.104,105
The active interaction of SFKs with FAK is essential for the integrin-mediated regulation of Rho GTPases.106 The small Rho GTPases Rho and Rac play major roles in regulating the actin cytoskeleton. Their activation is regulated by guanine nucleotide exchange factors that promote GDP release from the GTPases and subsequent GTP loading. Some of these nucleotide exchange factors are also substrates of FAK (as mentioned above).67,107 Rho GTPases play an important role in regulating HSC§ migration, proliferation and self-renewal. The ubiquitously expressed Rho GTPase Rac1, in particular, is essential in HSC§ homing and engraftment.108 The hematopoietic cell-specific Rho family member Rac2 is critical for HSC|| migration and adhesion. It also modulates Rac1 and Cdc42 activities.109,110 Cdc42 plays a role in HSC§ migration and retention in the niche.111 RhoA negatively regulates human HSC** migration. And, correspondingly, active RhoA supports the retention of HSCs** in their niche.112
Paxillin is recruited to integrin adhesion sites early on during integrin-mediated signalling.113 It is able to bind directly to the cytoplasmic tail of the integrin alpha4 chain114 and has several protein-interaction modules and phosphorylation sites. Phosphorylation regulates the interaction of paxillin with other proteins. Paxillin's main function is to mediate and orchestrate the binding of signalling molecules and cytoskeleton-linking molecules to the cytoplasmic domain of integrins, thereby contributing to the regulation of adhesion turnover and cell migration.69
Detailed analyses of the signalling pathways downstream of integrins in HSCs† are available for the process of chemokine-stimulated cell migration. During this process, phosphorylation of FAK, Pyk2, and paxillin as well as the adaptor molecules P130CAS (synonym: BCAR1), Crk and Crk-L is induced.115,116Fig. 2 summarizes the molecules that are known to be involved in integrin–actin-linkage and integrin-mediated signalling in HSCs.
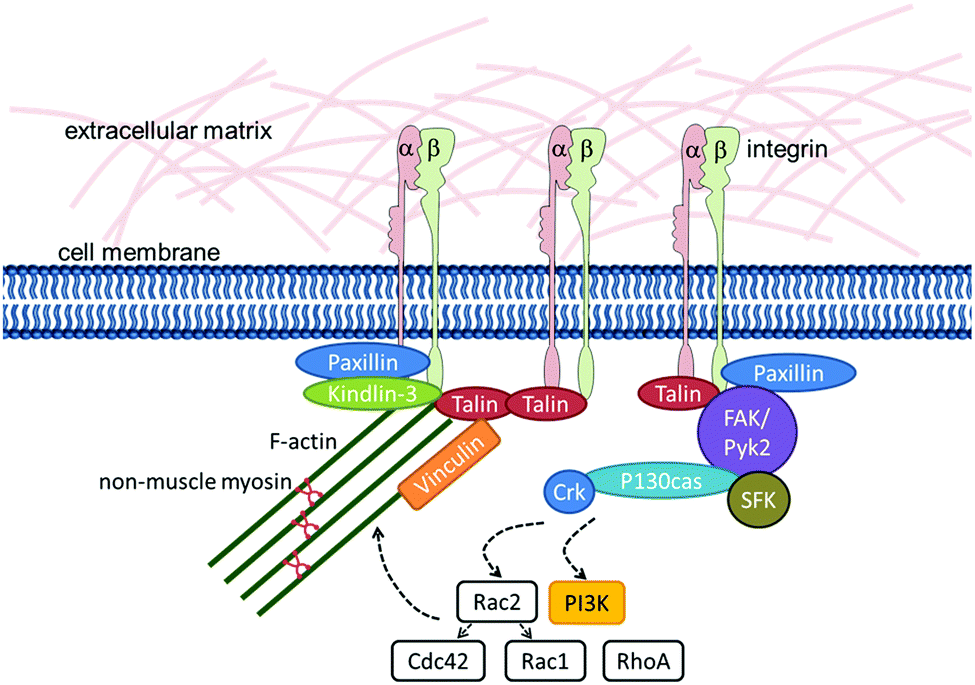 | ||
| Fig. 2 Integrin–actin interaction and the integrin-signalling complex. The illustration highlights the molecules downstream of integrins that are expressed by HSPCs and/or play a role in HSPC biology. | ||
Mechanical properties
Sensitivity to matrix stiffness
In their niches, stem cells experience microenvironments with specific mechanical properties. Research using polymeric materials with an adjustable E-modulus has revealed that both embryonic and adult stem cells are sensitive to substrate stiffness.117–120 Engler et al. were the first to report on the ability to control and direct the differentiation of mesenchymal stem cells by changing the E-modulus of the substrate. Interestingly, cellular differentiation reflected the matrix's stiffness. For example, on soft matrices (resembling the brain's mechanical properties) mesenchymal stem cells differentiated into cells of the neural lineage.117In 2010, Holst and co-workers reported that the proliferation of HSCs§,†† was increased when they cultured murine bone marrow cells or human mononuclear cells from umbilical cord on tropoelastin-coated substrates. Tropoelastin is the most flexible known biomolecule and its stretchability proved to be the pro-proliferative parameter in this experimental setup.53 Both HSC† adhesion and migration depend on the mechanical properties of the substrate, as we could show employing fibronectin-coated poly(ethylene glycol) (PEG) hydrogels. Our research showed that HSCs† adhered better and were more motile on stiffer hydrogels than on softer ones.54 Similarly, it has been reported that the elasticity of collagen hydrogels influences HSC morphology.52
The mechanical properties that stem cells experience in their stem cell niche are determined by the ECM and the adjacent niche cells. Osteoblasts, which represent a major cell type in the endosteal HSC niche, respond to adrenergic stimulation (as it occurs during mobilization of HSCs from their niche to the blood circulation) by changing their morphology – they flatten.43 And, as our research shows, they simultaneously change their mechanical properties by getting stiffer.54 This is a first hint suggesting that niche stiffness is not a static parameter, but rather a property that is dynamically regulated during physiological processes.
One example of changing tissue stiffness caused by physiological processes is the impact of ageing. In tissues like skin both the composition of the ECM and molecular cross-linking in the ECM change during ageing, resulting in matrix stiffening.121,122 These changes are considered cell-extrinsic as they are a result of the aging cellular environment. In addition, HSCs also undergo cell-intrinsic age-related modifications that lead to a decline in their functionality.123–125 These may lead to age-related changes in the mechanosensitivity of HSCs, as the small Rho GTPase Cdc42 was shown to play a central role in HSC ageing. Cdc42 activity is increased in aged HSCs and is associated with a loss of polarity in these cells.123,124 As Cdc42 acts downstream of integrins and is involved in the regulation of the actin cytoskeleton, it may also be involved in the mechanosensing processes downstream of integrins. Whether ageing impacts biophysical cues (including matrix stiffness and mechanosensing processes) in the hematopoietic compartment, however, is yet to be determined.
Mechanotransduction
As described in the previous chapter, HSCs are sensitive to the mechanical properties of their environment. But how do they transmit the mechanical signals provided by the environment to the cell's interior? A prerequisite for the cell's ability to sense the mechanical properties of substrates is the existence of forces acting between the cell and the substrate. In order to react to the physical stimulus that is elicited by the mechanical properties of the environment, the stimulus must be translated into a biochemical signal in the cell's interior which is (i) readable by the cell, and (ii) results in a direct response (e.g., change of morphology or, on a longer timescale, gene expression). When a cell interacts with an ECM with distinct mechanical properties, the cell will bind to its specific ligand through an integrin that, on the one hand, connects the ECM to the cytoskeleton and, on the other hand, elicits intracellular signalling. As described above, the resulting outside-in and inside-out signalling leads to local changes in the cytoskeleton. This results in the generation of internal forces that counteract the externally applied forces. This dynamic interplay of reciprocally pulling forces leads to a condition in which the cytoskeleton is under isometric tension, thereby stabilizing the cell morphology and enabling the cell to sense external forces and react appropriately.126–128 The direct mechanical cross-talk between integrins and the actin cytoskeleton (i) provides pulling forces that are a prerequisite for the successful development of adhesive structures and (ii) appears to be a key element in the mechanosensing of cells. Many molecules are involved in linking integrins to the actin cytoskeleton, regulating their dynamics and eliciting signalling cascades. At the same time, the assembly of this multiprotein complex is dependent on the generation of force at many different levels and steps. Therefore, it appears that it is not a single protein which acts as ‘the one and only’ mechanosensor. Instead, the entire multiprotein complex – including its molecular network that acts in response to applied and generated forces – has to be regarded as a mechanosensory unit.67 In the following, some molecules of this network that appear to be of particular importance in the mechanotransduction process of HSCs will be highlighted (Fig. 3).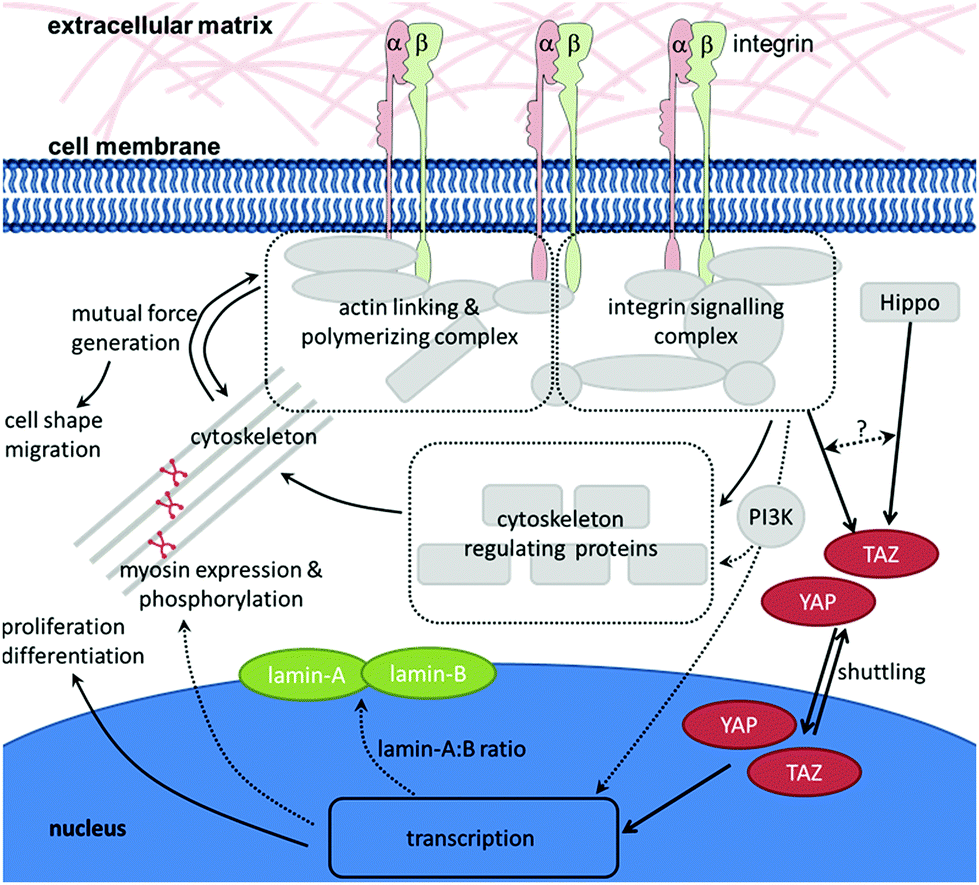 | ||
| Fig. 3 Mechanosensing pathways in HSCs. The drawing schematically depicts the mechanosensory units and molecules known to play a role in HSCs. The integrin adhesion and signalling complexes that act in response to externally applied and internally generated forces can be regarded as one mechanosensing unit that acts as a whole. Cellular signalling and structural factors like PI3K, YAP and TAZ, non-muscle myosin II as well as lamin-A and -B are involved in or regulated by mechanostransduction. Pathways that remain to be elucidated are indicated by dashed arrows, the question mark indicates that crosstalk between the Hippo and mechanotransduction signalling pathways has been proposed. | ||
Non-muscle myosin II is a central player in the generation of force in cells. Since Engler et al. first reported on the mechanosensitivity of mesenchymal stem cells, it has become generally accepted that non-muscle myosin II is involved in the mechanotransduction process of these cells. Pharmacological inhibition of non-muscle myosin II using blebbistatin has revealed its role in mechanosensing of mesenchymal stem cells117 as well as HSCs.§53 Non-muscle myosin II is composed of two heavy chains, two essential light chains, and two regulatory light chains. The heavy chains interact with the actin cytoskeleton and host the motor unit. The essential light chains act as stabilizing elements. The regulatory light chains contain several phosphorylation sites. Phosphorylation of these sites, e.g., by myosin light chain kinase (MLCK) or Rho-associated protein kinase (ROCK), contributes to the regulation of myosin II activity. The heavy chains carry additional phosphorylation sites, whose phosphorylation may influence myosin II activity even more directly.77 The expression level as well as phosphorylation pattern of non-muscle myosin II appear to depend on the stiffness of the underlying matrix.129 As mentioned earlier, HSCs† express both non-muscle myosin II isoforms A and B. While non-muscle myosin IIA is expressed throughout the entire hematopoietic differentiation, myosin IIB vanishes with differentiation. When cultured on stiff substrates (34 kPa, resembling the osteoid matrix of the bone endosteum) myosin IIB is polarized in the uropod of adhering, polarized HSCs. In contrast, on soft matrices that resemble bone marrow in their stiffness (0.3 kPa) myosin IIB polarization is not observed. Myosin IIB polarization contributes to the asymmetric cell division of HSCs. During this process, myosin IIB accumulates in the daughter cell which expresses higher levels of the HSPC marker CD34. Myosin IIA activity is enhanced on stiff matrices (more myosin IIA is dephosphorylated at S1943), whereas on soft matrices it is decreased (phosphorylation at S1943 increased).62 This suggests that non-muscle myosin II is a central element in mechanotransduction processes and biophysical regulation in anchorage-dependent cells and HSCs likewise.
Like myosin II, the signalling pathways downstream of integrins are also subjected to mechanosensitive signalling. We could show that phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) plays a role in those mechanotransduction processes downstream of integrins that regulate HSC† adherence and motility. When PI3K is specifically inhibited, adhesion and motility on stiff matrices are reduced down to a level similar to that observed on soft matrices.54 Which signals act up- and downstream of PI3K remains to be elucidated.
Another particularly interesting potential mechanosensor located downstream of integrins is the adaptor molecule P130CAS, which undergoes conformation changes under mechanical stretching. As it is being stretched phosphorylation sites are exposed and become available as substrates for interacting kinases of the Src family.130 Whether P130CAS in HSCs acts in a similar manner is the subject of further studies.
All in all, as HSC sensitivity to matrix stiffness has been a topic of investigation only during the last 4 years, very little is known on mechanotransduction in these cells. Most studies describe mechanosensitivity phenomenologically rather than elucidate the underlying molecular mechanisms. Therefore, exploring mechanotransduction in HSCs remains a hot topic for future investigation.
Transducing mechanical signals into the nucleus
Short-term effects of mechanical signals on HSCs include altered adhesion and migration54 whilst, on a longer timescale, HSCs react with changes in their proliferation and differentiation.53 Changes in protein conformation, complex composition and posttranslational modifications (such as phosphorylation) are sufficient for immediate responses to intracellular forces that are elicited by the mechanical properties of the environment. Long-term reactions, in contrast, necessitate modifications in gene expression. This requires that the mechanical signal is first translated into the cell's interior and then, in a second step, into the nucleus. Here, the arriving signal can lead to changes in the activity of transcription factors as well as the nuclear or cytoplasmic localization of molecules that ultimately affect gene expression.Such effects have been described for transcriptional cofactors YAP (yes-associated protein) and TAZ (transcriptional co-activator with PDZ binding motif; synonym: WWTR1).131,132 YAP and TAZ are closely related proteins that shuttle between the nucleus and the cytoplasm. Inside the nucleus they interact with transcription factors such as TEA domain family members. YAP and TAZ regulate organ size as well as adult stem cells.133–137 Both are downstream effectors of the Hippo pathway, the pathway that regulates YAP and TAZ localization and degradation.138 YAP and TAZ have been implicated as major effectors in mechanotransduction, because they communicate mechanically elicited signals into the nucleus, thereby affecting gene transcription. On stiff substrates YAP and TAZ are located inside the nucleus and are active, whereas on soft matrices YAP and TAZ are positioned in the cytoplasm and are functionally inactive.131 As force transduction is closely associated with the actomyosin cytoskeleton, YAP and TAZ activity depend on the integrity of the actin cytoskeleton and Rho activity (which regulates cytoskeletal dynamics).131,139–141 Crosstalk between the two known parallel-acting YAP and TAZ regulating cues – the Hippo pathway and cytoskeletal dynamics – has been proposed.132 Concerning its occurrence, YAP is detectable at low levels only in murine long-term HSCs (Lin− Sca1+ cKit+ Flt3− CD34−), but not in murine short-term HSCs (Lin− Sca1+ cKit+ Flt3− CD34+) or Lin+ hematopoietic lineages. Furthermore, YAP expression seems to not impact HSC function, which is in contrast to the role ascribed to YAP in other tissue-specific adult stem cells.142 Therefore, it appears questionable whether the results obtained for YAP concerning mechanotransduction in, e.g., mesenchymal stem cells also hold true for HSCs.
The nuclear lamina has been proposed as an additional element that regulates YAP translocation. Research on mesenchymal stem cells has shown the nucleoskeleton protein lamin-A to be more abundant in cells cultured on stiffer substrates than in those grown on softer substrates. Additionally, lamin-A conformation proved to be mechanosensitive.143 Lamin-A also coregulates key factors such as YAP and serum response protein (SRP).143 As lamins determine the mechanical properties of the nucleus, the relative abundance of the two isoforms lamin-A and -B plays an important role in transmigration processes of hematopoietic cells, including HSCs.†144 Transmigration can occur both during homeostasis and disease as well as after pharmacological induction. A small number of HSCs constantly leave their niche in the bone marrow, enter circulation by migrating across the endothelium of bone marrow sinusoids, circulate in the body, and return into the niche.145 The percentage of circulating HSCs can dramatically increase during alarm situations, such as infections or blood loss. In clinical settings (e.g., transplantation), mobilization of HSCs into the blood circulation can be induced in the donor through pharmacological treatment with cytokines such as G-CSF.146
Although some progress has been made in studying mechanotransduction, a detailed picture is lacking of how mechanical signals are transduced into the nucleus of HSCs (Fig. 3). Along the series of events that comprise the mechanotransduction signalling pathway in HSCs less and less is known about the components and events involved in passing along the signal the further downstream you look. Nevertheless, lessons learned from other cell types such as mesenchymal stem cells might provide useful information.
In addition to mechanical signals that create direct effects in HSCs (like matrix stiffness), indirect effects through neighboring niche cells that influence HSC behaviour and that are mechanosensitive (e.g., mesenchymal stem cells, osteoblasts, osteocytes and endothelial cells117,147–150) also seem likely. For example, stiffer matrices can induce differentiation of mesenchymal stem cells into the osteoblast lineage.117 This might, in turn, influence HSCs, as osteoblasts are known to provide signals to HSCs that are important in HSC maintenance in vivo§ in the niche8–10 and in vitro.†151 Osteocytes are subject to mechanical load in bone. In response to mechanical signals, they release signals that regulate osteoblast and osteoclast activity.148 Both osteoblasts and osteoclasts are involved in HSC niche formation in mice.44 Furthermore, osteocytes themselves are involved in the regulation of HSC mobilization in mice.46 Endothelial cells stiffen and contract in response to stiffer substrates, which facilitates transmigration of neutrophils through endothelial cell layers.150 Transmigration through vascular endothelia takes place during mobilization, homing and trafficking of HSCs. It would be interesting to investigate whether endothelial cells in the HSC microenvironment are also mechanosensitive and if the effects observed for neutrophil transmigration also hold true for HSC transmigration.
In conclusion, the direct as well as the indirect effects of matrix stiffness should be considered when trying to manipulate HSC behaviour through biophysical cues.
Nanopattern
One of the characteristic features of collagen type I fibrils is their banded structure with a periodicity of 67 nm, which can be observed using transmission electron and atomic force microscopy. The first three-dimensional model of collagen fibrils that accounted for the gap and overlap regions of monomers in the fibril that elicit the 67 nm banding was proposed in 1963.152,153 Since then, it has become evident that the ECM is highly organized and structured from the microscopic down to the nanometer scale. This means that a cell that interacts with the ECM receives – in addition to the chemical information provided by the molecule's amino acid sequence and composition – mechanical as well as (nano)structural and geometrical information. Cells are able to integrate these different signals and to respond specifically to nanostructural features of their environment.47,67 In order to investigate the impact of nanometer scale distances between specific adhesive ligands on cellular behaviour, nanopatterned substrates were developed that allow the positioning of ligands with precision in the nanometer range. Using block-copolymer micellar nanolithography, gold nanoparticles with a typical diameter of less than 10 nm were deposited on glass or silicium substrates in a quasi-hexagonal order. The lateral distance between individual gold nanoparticles can be adjusted between ∼20 and 300 nm, simply by altering the production parameters.154–156 Specific biofunctionalization of the gold nanoparticles is enforced by either passivating the area in between the gold nanoparticles by covalent attachment of PEG or by transferring the nanoparticle arrays to PEG hydrogels. Next, the gold nanoparticles are specifically functionalized via thiol chemistry with a biomolecule of choice.157,158RGD is a minimal integrin recognition motif that can be found in ECM molecules such as fibronectin. The RGD sequence in fibronectin is located in a loop structure.159 On artificial surfaces, a cyclic RGD peptide (cRGD) is usually employed, as it best mimics the amino acid sequence and the secondary structure of the naturally occurring integrin recognition motif. In our experiments with cRGD functionalized gold nanoparticle arrays, we could show that fully differentiated tissue cells such as fibroblasts and osteoblasts are highly sensitive to the nanostructured presentation of their ligands. Their adhesive and migratory behaviour as well as the assembly of focal adhesions are influenced by the ligand presentation on the nanometer scale.160–165 At lateral distances of 58 nm (and less) between the cRGD functionalized nanoparticles the cells adhered, spread out and developed mature focal adhesions. At distances of 73 nm and more, adhesion, spreading and focal adhesion development were impaired.160–162,164 The cells were also more motile on these substrates. Using substrates with lateral spacing gradients, it was shown that MC3T3 osteoblasts polarized and migrated towards smaller distances.163,165
Similarly, mesenchymal stem cell adhesion and differentiation are also influenced by nanoscale substrate features such as nanotopography and lateral spacing.166–170 For example, increasing the lateral distance between RGD ligands was shown to lead to diminished cell spreading and lower numbers of mature focal adhesions. At the same time, differentiation into the osteogenic lineage was impaired, while differentiation into adipocytes was increased.168 On nanotube arrays, small tube diameters promoted adhesion without differentiation, whereas cells on larger nanotubes were strongly elongated and differentiation into osteoblast-like cells was enhanced.169 On nanoscale gratings, mesenchymal stem cells aligned along the gratings and neuronal markers in these cells were up-regulated compared to neuronal marker expression in cells on flat substrates.170
The first report pointing at an influence of nanometer-sized features on HSCs described the enhancing effect of nanofibre scaffolds on the adhesion and proliferation of HSCs.†56 In the following years it became increasingly clear that, like anchorage-dependent cells, HSCs are influenced by the nanopatterning of their environment. We could show that the lateral distance between several ECM-derived ligands severely influenced the adhesive behaviour of HSCs.† Our findings show that the critical lateral distance between functionalized nanoparticles (i.e. a threshold value: at distances smaller than this value HSPCs are able to adhere, whereas at larger distances no adhesion occurs) is dependent on (i) cell type, (ii) the receptor that is targeted in the cell membrane and (iii) the presented ligand.57 For example, for the small ligand cRGD that is recognized by the integrin alpha5beta1 receptor of HSPCs the critical distance is ∼40 nm, whereas for a fibronectin protein domain (that also contains the RGD sequence and targets the same receptor in the membrane) the value lies between 85 nm and 110 nm. Explanations for the observed discrepancy between HSC adhesion to cRGD and the fibronectin domain are two-fold: (i) the fibronectin protein domain is larger and more flexible than the small cRGD peptide. This enables it to bridge larger distances (however, this cannot completely account for the substantial difference of ∼60 nm). (ii) The fibronectin domain carries not only the adhesive RGD sequence but also synergy sequences that stabilize binding to the integrin receptor. The increased binding stability should facilitate cell adhesion at lower ligand concentrations (in other words, binding at greater lateral distances between functionalized nanoparticles is possible).
In addition to influencing HSC† adhesion, nanopatterning also affects other cell functions such as gene expression,57 lipid raft clustering (which is assumed to be a prerequisite for successful signal transduction via transmembrane receptors), and receptor distribution in the cell.55
Little is known about the impact of nanotopography – meaning all sorts of nanoscale topological surface features – on HSCs. Despite the attention that the influence of nanotopography on mesenchymal stem cells has gained,166,167 such studies have not been performed with HSCs, probably because it was assumed that HSCs do not adhere strongly enough to materials surfaces to be able to sense nanotopographical features.171 However, studies using nanofiber scaffolds have shown that, in addition to biochemical surface functionalization, topographical surface texture influences HSC† adhesion and expansion. HSCs† adhered better and multiplied to higher cell numbers on nanofiber scaffolds than on flat surfaces.56,172 These findings support the idea that nanotopography, like nanopatterning, can indeed influence HSC behaviour.
Shear stress
HSCs develop in the aorta-gonad-mesonephros region during embryogenesis. During this process, shear stress elicited by blood flow is an important factor that impacts arising HSC precursors. Blood flow and the resulting shear stress even appear to be a prerequisite for successful HSC development.58,60 Studies have revealed that the influence of biomechanical forces on hematopoiesis commences during the embryonic stages. They also indicate that nitric oxide is a major mediator of shear stress-induced signalling in HSC precursors in both zebrafish and mice.58,60Adult HSCs in mammals are localized in the bone marrow and only small numbers circulate in the body.146 As they are circulating in the blood flow of mice, HSCs experience shear stresses at the vessel walls that in some regions exceed 600 dyne cm−2. In humans these shear stresses are lower by one order of magnitude.173 It has been shown in human HSCs† that shear stress impacts HSC adhesion and rolling behaviour on hyaluronan.174,175 However, most adult HSCs are found in bone marrow, where they might not be subjected to fluid flow directly. Nevertheless, other cells that are implicated in HSC niche functions do experience shear stress and could influence HSCs through paracrine signalling. Endothelial cells and also pericytes e.g., might be influenced by the relatively slow fluid flow in the vasculature that feeds the medullary cavities of bone. Furthermore, the fluid flow in the lacunar-canalicular network in compact bone (6–50 dyne cm−2) may, in addition to transporting nutrients and signalling molecules to osteocytes, also lead to their mechanical stimulation.59
All in all, the impact of shear stress on adult HSCs is still an open field. Even though fluid flow-induced shear stress may not play a major role inside the in vivo niche, it does play a role in the targeted in vitro/ex vivo proliferation and differentiation of HSCs.176,177 Successful HSC proliferation and differentiation ex vivo is an important goal of current research in the field of hematopoiesis and would have tremendous impact on possible clinical applications. Any kind of cell expansion on a sufficiently large scale for clinical applications inevitably requires the use of large bioreactors in order to ensure an adequate supply with medium containing essential nutrients and growth factors for a growing cell population. Fluid flow is an intrinsic parameter of such bioreactors. In order to be able to control and predict HSC behaviour in bioreactors, investigations on how fluid flow-induced shear stress influences HSC behaviour, including proliferation and differentiation, are needed.
3D versus 2D
In vivo HSCs are located in a three-dimensional environment. Therefore, culturing HSCs on flat tissue-culture dishes or in suspension is a highly artificial situation for these cells and is insufficient for realistically simulating the natural in vivo situation.29 One example of what may be missing in 2D cell culture systems is autocrine and paracrine signalling. These signals are important regulating mechanisms during hematopoiesis that act locally on short distances – either by acting back on the signalling cell itself (autocrine) or between adjacent cells/cells in close proximity (paracrine).178 In standard cell culture systems with relatively large amounts of medium these signals are rapidly diluted upon release and, therefore, cannot act locally. In contrast, in three-dimensional systems the effects of local accumulation (by release) or depletion (by consumption) of diffusible factors can be reproduced.Similarly, the diffusion of other soluble factors like nutrients or oxygen in the natural ECM leads to gradients that play an important role in many cellular processes such as migration or homing.179 These gradients can be mimicked more realistically by diffusion-limited 3D cell culture systems than standard 2D cell cultures.
Differences between the 2D and 3D culture systems include the distribution of soluble and diffusible factors as well as the way the ECM embeds the cells from all sides, rather than just providing one-sided contact. Providing a surrounding matrix leads to changes in the cells’ adhesive behaviour, morphology and migration. It has been shown that the induction of certain morphologies is an important determinant towards driving the differentiation of mesenchymal stem cells.180 This suggests that morphological changes induced by 2D cultures may also influence stem cell function. However, HSCs and their direct progenies (i.e. early hematopoietic progenitors) all have a similar round shape. Therefore, it seems unlikely that the concept of morphology that impacts differentiation can be transferred directly from mesenchymal to hematopoietic cells.
Nevertheless, it has become clear that cellular answers to substrate-bound as well as soluble signals differ in 2D and 3D systems. Therefore, an important future task will be to study a wide range of parameters in 3D systems that resemble the natural stem cell niches, including the influence of ECM-molecules, cell–cell interaction (contact and communication) and growth factors as well as biophysical signals (such as spatial distribution of signals on the micro- and nanoscale) and mechanical parameters.
Several different approaches to culture and investigate HSCs in vitro using 3D systems have been tested up until now: HSCs†,§ have been encapsulated in hydrogels made of natural (e.g., collagen, hyaluronic acid) or synthetic (e.g., polyacrylates) monomers;48,50,52 Other approaches included seeding of HSCs† onto scaffolds made of fibres or fibre meshworks,51 as well as culturing HSCs†,‡‡ in macroporous scaffolds that resemble the spongy architecture of trabecular bones49,181–183 (these host the red, blood-forming bone marrow); Furthermore, microwell-arrays have been used as quasi- or pseudo-3D environments for HSCs§§,¶¶ in order to delineate signals and parameters that are present in the HSC niche.184–187 All of these studies showed that HSCs respond and act differently in 3D materials than in standard 2D culture, backing the assumption that conventional 2D culture is a highly artificial situation for HSCs, which may even induce ‘unnatural’ cell behaviour. Using RGD-functionalized macroporous PEG scaffolds we determined that the support of HSCs† by feeder cells is more pronounced in 3D than on 2D scaffolds. Furthermore, only when combining 3D scaffolds and feeder cell support did more than 90% of the cells preserve CD34 marker expression throughout culture. Neither stromal support in 2D nor 3D scaffolds alone was able to support HSC marker maintenance to this degree.49
Each of the afore-mentioned approaches for HSC 3D culture has its advantages and disadvantages – and each system can best imitate a different aspect of the natural 3D situation. A combination of the different approaches and techniques might be a promising future approach to mimic the natural niche as realistically as possible. Such artificial niches may be useful as model systems for fundamental research on HSC niches both in health and disease. Furthermore, they could be applied in drug screening systems, minimizing the need for animal testing. Last but not least, they are promising targets in the field of biomaterial-based HSC expansion for clinical use.
Conclusions
Although after more than 50 years of research studying blood-forming stem cells HSCs are clearly the best studied adult stem cells today, surprisingly little is known about the biophysical regulation of these cells. More and more data suggest that HSCs are sensitive to physical signals. This allows the conclusion that to be able to direct HSC behaviour all types of environmental stimuli that act on the cell – biological, chemical as well as physical – must be taken into account. Guiding the behaviour and functions of HSCs by engineered materials is a difficult but promising future challenge. However, before being able to design such tailor-made materials, fundamental questions on the biophysical regulation of HSCs remain to be answered.Acknowledgements
The authors thank Nina Grunze for careful editing of the manuscript. C.L.-T. acknowledges support by the BMBF NanoMatFutur programme (FKZ: 13N12968), the BioInterfaces programme of the Helmholtz-Association and the ‘Käthe und Josef Klinz-Stiftung’. This work is also part of the excellence cluster CellNetworks at the University of Heidelberg. J.P.S. has a Weston Visiting Professorship at the Weizmann Institute of Science. The Max Planck Society is highly acknowledged for its support.Notes and references
- T. Graf and M. Stadtfeld, Cell Stem Cell, 2008, 3, 480–483 CrossRef CAS PubMed.
- M. Ogawa, Blood, 1993, 81, 2844–2853 CAS.
- E. D. Thomas and K. G. Blume, Biol. Blood Marrow Transplant., 1999, 5, 341–346 CrossRef CAS.
- A. Dahlberg, C. Delaney and I. D. Bernstein, Blood, 2011, 117, 6083–6090 CrossRef CAS PubMed.
- R. Schofield, Blood Cells, 1978, 4, 7–25 CAS.
- S. J. Morrison and D. T. Scadden, Nature, 2014, 505, 327–334 CrossRef CAS PubMed.
- D. T. Scadden, Nature, 2006, 441, 1075–1079 Search PubMed.
- L. M. Calvi, G. B. Adams, K. W. Weibrecht, J. M. Weber, D. P. Olson, M. C. Knight, R. P. Martin, E. Schipani, P. Divieti, F. R. Bringhurst, L. A. Milner, H. M. Kronenberg and D. T. Scadden, Nature, 2003, 425, 841–846 Search PubMed.
- F. Arai, A. Hirao, M. Ohmura, H. Sato, S. Matsuoka, K. Takubo, K. Ito, G. Y. Koh and T. Suda, Cell, 2004, 118, 149–161 CrossRef CAS PubMed.
- J. Zhang, C. Niu, L. Ye, H. Huang, X. He, W. G. Tong, J. Ross, J. Haug, T. Johnson, J. Q. Feng, S. Harris, L. M. Wiedemann, Y. Mishina and L. Li, Nature, 2003, 425, 836–841 CrossRef CAS PubMed.
- M. J. Kiel, O. H. Yilmaz, T. Iwashita, O. H. Yilmaz, C. Terhorst and S. J. Morrison, Cell, 2005, 121, 1109–1121 CrossRef CAS PubMed.
- Y. Kunisaki, I. Bruns, C. Scheiermann, J. Ahmed, S. Pinho, D. Zhang, T. Mizoguchi, Q. Wei, D. Lucas, K. Ito, J. C. Mar, A. Bergman and P. S. Frenette, Nature, 2013, 502, 637–643 CrossRef CAS PubMed.
- L. Ding and S. J. Morrison, Nature, 2013, 495, 231–235 CrossRef CAS PubMed.
- A. Ehninger and A. Trumpp, J. Exp. Med., 2011, 208, 421–428 CrossRef CAS PubMed.
- S. Doulatov, F. Notta, E. Laurenti and J. E. Dick, Cell Stem Cell, 2012, 10, 120–136 Search PubMed.
- R. Peerani and P. W. Zandstra, J. Clin. Invest., 2010, 120, 60–70 Search PubMed.
- M. Votteler, P. J. Kluger, H. Walles and K. Schenke-Layland, Macromol. Biosci., 2010, 10, 1302–1315 CrossRef CAS PubMed.
- B. M. Sagar, S. Rentala, P. N. Gopal, S. Sharma and A. Mukhopadhyay, Biochem. Biophys. Res. Commun., 2006, 350, 1000–1005 CrossRef CAS PubMed.
- U. Siler, M. Seiffert, S. Puch, A. Richards, B. Torok-Storb, C. A. Muller, L. Sorokin and G. Klein, Blood, 2000, 96, 4194–4203 CAS.
- T. Yokota, K. Oritani, H. Mitsui, K. Aoyama, J. Ishikawa, H. Sugahara, I. Matsumura, S. Tsai, Y. Tomiyama, Y. Kanakura and Y. Matsuzawa, Blood, 1998, 91, 3263–3272 CAS.
- S. K. Nilsson, H. M. Johnston, G. A. Whitty, B. Williams, R. J. Webb, D. T. Denhardt, I. Bertoncello, L. J. Bendall, P. J. Simmons and D. N. Haylock, Blood, 2005, 106, 1232–1239 CrossRef CAS PubMed.
- S. Stier, Y. Ko, R. Forkert, C. Lutz, T. Neuhaus, E. Grunewald, T. Cheng, D. Dombkowski, L. M. Calvi, S. R. Rittling and D. T. Scadden, J. Exp. Med., 2005, 201, 1781–1791 CrossRef CAS PubMed.
- S. Badylak, T. Gilbert and J. Myers-Irvin, in Tissue Eng., ed. C. v. Blitterswijk, P. Thomsen, A. Lindahl, J. Hubbell, D. F. Williams, R. Cancedda, J. D. d. Bruijn and J. Sohier, Academic Press, Burlington, 2008, pp. 121–143 Search PubMed.
- D. N. Haylock and S. K. Nilsson, Regen. Med., 2006, 1, 437–445 CrossRef CAS PubMed.
- S. K. Nilsson, M. E. Debatis, M. S. Dooner, J. A. Madri, P. J. Quesenberry and P. S. Becker, J. Histochem. Cytochem., 1998, 46, 371–377 CrossRef CAS PubMed.
- M. Hines, L. Nielsen and J. Cooper-White, J. Chem. Technol. Biotechnol., 2008, 83, 421–443 Search PubMed.
- G. Klein, Experientia, 1995, 51, 914–926 CrossRef CAS.
- Y. Gu, L. Sorokin, M. Durbeej, T. Hjalt, J. I. Jonsson and M. Ekblom, Blood, 1999, 93, 2533–2542 CAS.
- B. M. Baker and C. S. Chen, J. Cell Sci., 2012, 125, 3015–3024 CrossRef CAS PubMed.
- M. Ekblom, R. Fassler, B. Tomasini-Johansson, K. Nilsson and P. Ekblom, J. Cell Biol., 1993, 123, 1037–1045 CrossRef CAS.
- Y. C. Gu, J. F. Talts, D. Gullberg, R. Timpl and M. Ekblom, Eur. J. Haematol., 2001, 67, 176–184 Search PubMed.
- S. P. Hergeth, W. K. Aicher, M. Essl, T. D. Schreiber, T. Sasaki and G. Klein, Exp. Hematol., 2008, 36, 1022–1034 Search PubMed.
- J. R. Fraser, T. C. Laurent and U. B. Laurent, J. Intern. Med., 1997, 242, 27–33 Search PubMed.
- S. K. Nilsson, D. N. Haylock, H. M. Johnston, T. Occhiodoro, T. J. Brown and P. J. Simmons, Blood, 2003, 101, 856–862 CrossRef CAS PubMed.
- J. Grassinger, D. N. Haylock, M. J. Storan, G. O. Haines, B. Williams, G. A. Whitty, A. R. Vinson, C. L. Be, S. Li, E. S. Sorensen, P. P. Tam, D. T. Denhardt, D. Sheppard, P. F. Choong and S. K. Nilsson, Blood, 2009, 114, 49–59 Search PubMed.
- S. Mendez-Ferrer, T. V. Michurina, F. Ferraro, A. R. Mazloom, B. D. Macarthur, S. A. Lira, D. T. Scadden, A. Ma'ayan, G. N. Enikolopov and P. S. Frenette, Nature, 2010, 466, 829–834 CrossRef CAS PubMed.
- Y. Omatsu, T. Sugiyama, H. Kohara, G. Kondoh, N. Fujii, K. Kohno and T. Nagasawa, Immunity, 2010, 33, 387–399 CrossRef CAS PubMed.
- T. Sugiyama, H. Kohara, M. Noda and T. Nagasawa, Immunity, 2006, 25, 977–988 CrossRef CAS PubMed.
- S. Yamazaki, H. Ema, G. Karlsson, T. Yamaguchi, H. Miyoshi, S. Shioda, M. M. Taketo, S. Karlsson, A. Iwama and H. Nakauchi, Cell, 2011, 147, 1146–1158 CrossRef CAS PubMed.
- A. Chow, D. Lucas, A. Hidalgo, S. Mendez-Ferrer, D. Hashimoto, C. Scheiermann, M. Battista, M. Leboeuf, C. Prophete, N. van Rooijen, M. Tanaka, M. Merad and P. S. Frenette, J. Exp. Med., 2011, 208, 261–271 CrossRef CAS PubMed.
- M. J. Christopher, M. Rao, F. Liu, J. R. Woloszynek and D. C. Link, J. Exp. Med., 2011, 208, 251–260 CrossRef CAS PubMed.
- I. G. Winkler, N. A. Sims, A. R. Pettit, V. Barbier, B. Nowlan, F. Helwani, I. J. Poulton, N. van Rooijen, K. A. Alexander, L. J. Raggatt and J. P. Levesque, Blood, 2010, 116, 4815–4828 CrossRef CAS PubMed.
- Y. Katayama, M. Battista, W. M. Kao, A. Hidalgo, A. J. Peired, S. A. Thomas and P. S. Frenette, Cell, 2006, 124, 407–421 CrossRef CAS PubMed.
- A. Mansour, G. Abou-Ezzi, E. Sitnicka, S. E. Jacobsen, A. Wakkach and C. Blin-Wakkach, J. Exp. Med., 2012, 209, 537–549 CrossRef CAS PubMed.
- A. Mansour, A. Wakkach and C. Blin-Wakkach, Cell Cycle, 2012, 11, 2045–2046 Search PubMed.
- N. Asada, Y. Katayama, M. Sato, K. Minagawa, K. Wakahashi, H. Kawano, Y. Kawano, A. Sada, K. Ikeda, T. Matsui and M. Tanimoto, Cell Stem Cell, 2013, 12, 737–747 CrossRef CAS PubMed.
- C. Lee-Thedieck and J. P. Spatz, Macromol. Rapid Commun., 2012, 33, 1432–1438 Search PubMed.
- I. Leisten, R. Kramann, M. S. Ventura Ferreira, M. Bovi, S. Neuss, P. Ziegler, W. Wagner, R. Knuchel and R. K. Schneider, Biomaterials, 2012, 33, 1736–1747 CrossRef CAS PubMed.
- A. Raic, L. Rodling, H. Kalbacher and C. Lee-Thedieck, Biomaterials, 2014, 35, 929–940 CrossRef CAS PubMed.
- M. S. Ventura Ferreira, W. Jahnen-Dechent, N. Labude, M. Bovi, T. Hieronymus, M. Zenke, R. K. Schneider and S. Neurs, Biomaterials, 2012, 33, 6987–6997 CrossRef CAS PubMed.
- Q. Feng, C. Chai, X. S. Jiang, K. W. Leong and H. Q. Mao, J. Biomed. Mater. Res., Part A, 2006, 78, 781–791 CrossRef PubMed.
- J. S. Choi and B. A. Harley, Biomaterials, 2012, 33, 4460–4468 CrossRef CAS PubMed.
- J. Holst, S. Watson, M. S. Lord, S. S. Eamegdool, D. V. Bax, L. B. Nivison-Smith, A. Kondyurin, L. Ma, A. F. Oberhauser, A. S. Weiss and J. E. Rasko, Nat. Biotechnol., 2010, 28, 1123–1128 CrossRef CAS PubMed.
- C. Lee-Thedieck, N. Rauch, R. Fiammengo, G. Klein and J. P. Spatz, J. Cell Sci., 2012, 125, 3765–3775 CrossRef CAS PubMed.
- E. Altrock, C. A. Muth, G. Klein, J. P. Spatz and C. Lee-Thedieck, Biomaterials, 2012, 33, 3107–3118 CrossRef CAS PubMed.
- K. N. Chua, C. Chai, P. C. Lee, Y. N. Tang, S. Ramakrishna, K. W. Leong and H. Q. Mao, Biomaterials, 2006, 27, 6043–6051 Search PubMed.
- C. A. Muth, C. Steinl, G. Klein and C. Lee-Thedieck, PLoS One, 2013, 8, e54778 Search PubMed.
- L. Adamo, O. Naveiras, P. L. Wenzel, S. McKinney-Freeman, P. J. Mack, J. Gracia-Sancho, A. Suchy-Dicey, M. Yoshimoto, M. W. Lensch, M. C. Yoder, G. Garcia-Cardena and G. Q. Daley, Nature, 2009, 459, 1131–1135 CrossRef CAS PubMed.
- H. J. Lee, N. Li, S. M. Evans, M. F. Diaz and P. L. Wenzel, Differentiation, 2013, 86, 92–103 CrossRef CAS PubMed.
- T. E. North, W. Goessling, M. Peeters, P. Li, C. Ceol, A. M. Lord, G. J. Weber, J. Harris, C. C. Cutting, P. Huang, E. Dzierzak and L. I. Zon, Cell, 2009, 137, 736–748 CrossRef CAS PubMed.
- D. Van der Velde-Zimmermann, M. A. Verdaasdonk, L. H. Rademakers, R. A. De Weger, J. G. Van den Tweel and P. Joling, Exp. Cell Res., 1997, 230, 111–120 CrossRef CAS PubMed.
- J. W. Shin, A. Buxboim, K. R. Spinler, J. Swift, D. A. Christian, C. A. Hunter, C. Leon, C. Gachet, P. C. Dingal, I. L. Ivanovska, F. Rehfeldt, J. A. Chasis and D. E. Discher, Cell Stem Cell, 2014, 14, 81–93 CrossRef CAS PubMed.
- T. Ohashi, Y. Ishii, Y. Ishikawa, T. Matsumoto and M. Sato, Biomed. Mater. Eng., 2002, 12, 319–327 CAS.
- A. J. Engler, F. Rehfeldt, S. Sen and D. E. Discher, Methods Cell Biol., 2007, 83, 521–545 CAS.
- A. J. Engler, L. Richert, J. Y. Wong, C. Picart and D. E. Discher, Surf. Sci., 2004, 570, 142–154 CrossRef CAS PubMed.
- A. Buxboim, K. Rajagopal, A. E. X. Brown and D. E. Discher, J. Phys.: Condens. Matter, 2010, 22 Search PubMed.
- B. Geiger, J. P. Spatz and A. D. Bershadsky, Nat. Rev. Mol. Cell Biol., 2009, 10, 21–33 CrossRef CAS PubMed.
- B. M. Gumbiner, Cell, 1996, 84, 345–357 CrossRef CAS.
- D. S. Harburger and D. A. Calderwood, J. Cell Sci., 2009, 122, 159–163 CrossRef CAS PubMed.
- R. O. Hynes, Cell, 2002, 110, 673–687 CrossRef CAS.
- J. D. Humphries, A. Byron and M. J. Humphries, J. Cell Sci., 2006, 119, 3901–3903 CrossRef CAS PubMed.
- D. A. Calderwood, S. J. Shattil and M. H. Ginsberg, J. Biol. Chem., 2000, 275, 22607–22610 CrossRef CAS PubMed.
- E. A. Evans and D. A. Calderwood, Science, 2007, 316, 1148–1153 CrossRef CAS PubMed.
- D. A. Calderwood, J. Cell Sci., 2004, 117, 657–666 CrossRef CAS PubMed.
- B. Geiger, A. Bershadsky, R. Pankov and K. M. Yamada, Nat. Rev. Mol. Cell Biol., 2001, 2, 793–805 CrossRef CAS PubMed.
- C. K. Choi, M. Vicente-Manzanares, J. Zareno, L. A. Whitmore, A. Mogilner and A. R. Horwitz, Nat. Cell Biol., 2008, 10, 1039–1050 CrossRef CAS PubMed.
- J. W. Shin, J. Swift, I. Ivanovska, K. R. Spinler, A. Buxboim and D. E. Discher, Differentiation, 2013, 86, 77–86 CrossRef CAS PubMed.
- C. Decraene, L. Garcon, C. Lacout, S. Sabri, C. Auffray, W. Vainchenker, D. Dumenil, G. Pietu and F. Svinartchuk, Biochem. Biophys. Res. Commun., 2004, 318, 439–443 Search PubMed.
- R. Zaidel-Bar, C. Ballestrem, Z. Kam and B. Geiger, J. Cell Sci., 2003, 116, 4605–4613 CrossRef CAS PubMed.
- A. V. Fonseca and D. Corbeil, Commun. Integr. Biol., 2011, 4, 201–204 CrossRef CAS PubMed.
- A. V. Fonseca, D. Freund, M. Bornhauser and D. Corbeil, J. Biol. Chem., 2010, 285, 31661–31671 Search PubMed.
- B. Giebel, D. Corbeil, J. Beckmann, J. Hohn, D. Freund, K. Giesen, J. Fischer, G. Kogler and P. Wernet, Blood, 2004, 104, 2332–2338 CrossRef CAS PubMed.
- F. Ye, C. Kim and M. H. Ginsberg, Blood, 2012, 119, 26–33 CrossRef CAS PubMed.
- Y. Q. Ma, J. Qin, C. Wu and E. F. Plow, J. Cell Biol., 2008, 181, 439–446 CrossRef CAS PubMed.
- E. Montanez, S. Ussar, M. Schifferer, M. Bosl, R. Zent, M. Moser and R. Fassler, Genes Dev., 2008, 22, 1325–1330 CrossRef CAS PubMed.
- M. Moser, B. Nieswandt, S. Ussar, M. Pozgajova and R. Fassler, Nat. Med., 2008, 14, 325–330 CrossRef CAS PubMed.
- S. Tadokoro, S. J. Shattil, K. Eto, V. Tai, R. C. Liddington, J. M. de Pereda, M. H. Ginsberg and D. A. Calderwood, Science, 2003, 302, 103–106 CrossRef CAS PubMed.
- X. Zhang, G. Jiang, Y. Cai, S. J. Monkley, D. R. Critchley and M. P. Sheetz, Nat. Cell Biol., 2008, 10, 1062–1068 CrossRef CAS PubMed.
- T. Ohmori, Y. Kashiwakura, A. Ishiwata, S. Madoiwa, J. Mimuro, Y. Furukawa and Y. Sakata, J. Biol. Chem., 2010, 285, 31763–31773 CrossRef CAS PubMed.
- M. Moser, M. Bauer, S. Schmid, R. Ruppert, S. Schmidt, M. Sixt, H. V. Wang, M. Sperandio and R. Fassler, Nat. Med., 2009, 15, 300–305 CrossRef CAS PubMed.
- S. Ussar, H. V. Wang, S. Linder, R. Fassler and M. Moser, Exp. Cell Res., 2006, 312, 3142–3151 CrossRef CAS PubMed.
- C. G. Galbraith, K. M. Yamada and M. P. Sheetz, J. Cell Biol., 2002, 159, 695–705 CrossRef CAS PubMed.
- J. D. Humphries, P. Wang, C. Streuli, B. Geiger, M. J. Humphries and C. Ballestrem, J. Cell Biol., 2007, 179, 1043–1057 CrossRef CAS PubMed.
- I. B. Mazo, S. Massberg and U. H. von Andrian, Trends Immunol., 2011, 32, 493–503 CrossRef CAS PubMed.
- U. Steidl, S. Bork, S. Schaub, O. Selbach, J. Seres, M. Aivado, T. Schroeder, U. P. Rohr, R. Fenk, S. Kliszewski, C. Maercker, P. Neubert, S. R. Bornstein, H. L. Haas, G. Kobbe, D. G. Tenen, R. Haas and R. Kronenwett, Blood, 2004, 104, 81–88 CrossRef CAS PubMed.
- P. Roca-Cusachs, A. del Rio, E. Puklin-Faucher, N. C. Gauthier, N. Biais and M. P. Sheetz, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E1361–E1370 CrossRef CAS PubMed.
- R. Zaidel-Bar, S. Itzkovitz, A. Ma'ayan, R. Iyengar and B. Geiger, Nat. Cell Biol., 2007, 9, 858–867 CrossRef CAS PubMed.
- S. K. Mitra, D. A. Hanson and D. D. Schlaepfer, Nat. Rev. Mol. Cell Biol., 2005, 6, 56–68 CrossRef CAS PubMed.
- H. C. Chen, P. A. Appeddu, J. T. Parsons, J. D. Hildebrand, M. D. Schaller and J. L. Guan, J. Biol. Chem., 1995, 270, 16995–16999 CrossRef CAS PubMed.
- I. Hayashi, K. Vuori and R. C. Liddington, Nat. Struct. Biol., 2002, 9, 101–106 CrossRef CAS PubMed.
- K. Tachibana, T. Sato, N. D'Avirro and C. Morimoto, J. Exp. Med., 1995, 182, 1089–1099 CrossRef CAS.
- R. W. Tilghman and J. T. Parsons, Semin. Cancer Biol., 2008, 18, 45–52 Search PubMed.
- S. Melikova, S. J. Dylla and C. M. Verfaillie, Exp. Hematol., 2004, 32, 1051–1056 CrossRef CAS PubMed.
- J. Borneo, V. Munugalavadla, E. C. Sims, S. Vemula, C. M. Orschell, M. Yoder and R. Kapur, Exp. Hematol., 2007, 35, 1026–1037 CrossRef CAS PubMed.
- C. M. Orschell, J. Borneo, V. Munugalavadla, P. Ma, E. Sims, B. Ramdas, M. C. Yoder and R. Kapur, Exp. Hematol., 2008, 36, 655–666 CrossRef CAS PubMed.
- S. Huveneers and E. H. Danen, J. Cell Sci., 2009, 122, 1059–1069 CrossRef CAS PubMed.
- K. Burridge and K. Wennerberg, Cell, 2004, 116, 167–179 Search PubMed.
- Y. Gu, M. D. Filippi, J. A. Cancelas, J. E. Siefring, E. P. Williams, A. C. Jasti, C. E. Harris, A. W. Lee, R. Prabhakar, S. J. Atkinson, D. J. Kwiatkowski and D. A. Williams, Science, 2003, 302, 445–449 CrossRef CAS PubMed.
- F. C. Yang, S. J. Atkinson, Y. Gu, J. B. Borneo, A. W. Roberts, Y. Zheng, J. Pennington and D. A. Williams, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5614–5618 CrossRef CAS PubMed.
- M. Jansen, F. C. Yang, J. A. Cancelas, J. R. Bailey and D. A. Williams, Stem Cells, 2005, 23, 335–346 Search PubMed.
- L. Yang, L. Wang, H. Geiger, J. A. Cancelas, J. Mo and Y. Zheng, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5091–5096 Search PubMed.
- B. G. Jaganathan, F. Anjos-Afonso, A. Kumar and D. Bonnet, J. Biomed. Sci., 2013, 20, 66 CrossRef CAS PubMed.
- N. O. Deakin and C. E. Turner, J. Cell Sci., 2008, 121, 2435–2444 CrossRef CAS PubMed.
- C. Kummer and M. H. Ginsberg, Biochem. Pharmacol., 2006, 72, 1460–1468 CrossRef CAS PubMed.
- J. F. Wang, I. W. Park and J. E. Groopman, Blood, 2000, 95, 2505–2513 CAS.
- X. F. Zhang, J. F. Wang, E. Matczak, J. A. Proper and J. E. Groopman, Blood, 2001, 97, 3342–3348 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- N. D. Evans, C. Minelli, E. Gentleman, V. LaPointe, S. N. Patankar, M. Kallivretaki, X. Chen, C. J. Roberts and M. M. Stevens, Eur. Cell. Mater., 2009, 18, 1–13 CAS.
- P. M. Gilbert, K. L. Havenstrite, K. E. Magnusson, A. Sacco, N. A. Leonardi, P. Kraft, N. K. Nguyen, S. Thrun, M. P. Lutolf and H. M. Blau, Science, 2010, 329, 1078–1081 CrossRef CAS PubMed.
- N. Huebsch, P. R. Arany, A. S. Mao, D. Shvartsman, O. A. Ali, S. A. Bencherif, J. Rivera-Feliciano and D. J. Mooney, Nat. Mater., 2010, 9, 518–526 CrossRef CAS PubMed.
- D. T. Butcher, T. Alliston and V. M. Weaver, Nat. Rev. Cancer, 2009, 9, 108–122 CrossRef CAS PubMed.
- C. Frantz, K. M. Stewart and V. M. Weaver, J. Cell Sci., 2010, 123, 4195–4200 CrossRef CAS PubMed.
- C. Carrillo-Garcia and V. Janzen, Cell Stem Cell, 2012, 10, 481–482 CrossRef CAS PubMed.
- M. C. Florian, K. Dorr, A. Niebel, D. Daria, H. Schrezenmeier, M. Rojewski, M. D. Filippi, A. Hasenberg, M. Gunzer, K. Scharffetter-Kochanek, Y. Zheng and H. Geiger, Cell Stem Cell, 2012, 10, 520–530 CrossRef CAS PubMed.
- A. Kohler, V. Schmithorst, M. D. Filippi, M. A. Ryan, D. Daria, M. Gunzer and H. Geiger, Blood, 2009, 114, 290–298 CrossRef PubMed.
- A. Mammoto and D. E. Ingber, Curr. Opin. Cell Biol., 2009, 21, 864–870 CrossRef CAS PubMed.
- M. A. Schwartz, Cold Spring Harbor Perspect. Biol., 2010, 2, a005066 CAS.
- V. Vogel and M. Sheetz, Nat. Rev. Mol. Cell Biol., 2006, 7, 265–275 CrossRef CAS PubMed.
- M. Raab, J. Swift, P. C. Dingal, P. Shah, J. W. Shin and D. E. Discher, J. Cell Biol., 2012, 199, 669–683 Search PubMed.
- Y. Sawada, M. Tamada, B. J. Dubin-Thaler, O. Cherniavskaya, R. Sakai, S. Tanaka and M. P. Sheetz, Cell, 2006, 127, 1015–1026 CrossRef CAS PubMed.
- S. Dupont, L. Morsut, M. Aragona, E. Enzo, S. Giulitti, M. Cordenonsi, F. Zanconato, J. Le Digabel, M. Forcato, S. Bicciato, N. Elvassore and S. Piccolo, Nature, 2011, 474, 179–183 CrossRef CAS PubMed.
- G. Halder, S. Dupont and S. Piccolo, Nat. Rev. Mol. Cell Biol., 2012, 13, 591–600 CrossRef CAS PubMed.
- F. D. Camargo, S. Gokhale, J. B. Johnnidis, D. Fu, G. W. Bell, R. Jaenisch and T. R. Brummelkamp, Curr. Biol., 2007, 17, 2054–2060 CrossRef CAS PubMed.
- X. Cao, S. L. Pfaff and F. H. Gage, Genes Dev., 2008, 22, 3320–3334 Search PubMed.
- G. Halder and R. L. Johnson, Development, 2011, 138, 9–22 CrossRef CAS PubMed.
- M. Ramalho-Santos, S. Yoon, Y. Matsuzaki, R. C. Mulligan and D. A. Melton, Science, 2002, 298, 597–600 Search PubMed.
- B. Zhao, L. Li, Q. Lei and K. L. Guan, Genes Dev., 2010, 24, 862–874 CrossRef CAS PubMed.
- W. Hong and K. L. Guan, Semin. Cell Dev. Biol., 2012, 23, 785–793 CrossRef CAS PubMed.
- L. Sansores-Garcia, W. Bossuyt, K. Wada, S. Yonemura, C. Tao, H. Sasaki and G. Halder, EMBO J., 2011, 30, 2325–2335 CrossRef CAS PubMed.
- K. Wada, K. Itoga, T. Okano, S. Yonemura and H. Sasaki, Development, 2011, 138, 3907–3914 CrossRef CAS PubMed.
- B. Zhao, L. Li, L. Wang, C. Y. Wang, J. Yu and K. L. Guan, Genes Dev., 2012, 26, 54–68 CrossRef CAS PubMed.
- L. Jansson and J. Larsson, PLoS One, 2012, 7, e32013 Search PubMed.
- J. Swift, I. L. Ivanovska, A. Buxboim, T. Harada, P. C. Dingal, J. Pinter, J. D. Pajerowski, K. R. Spinler, J. W. Shin, M. Tewari, F. Rehfeldt, D. W. Speicher and D. E. Discher, Science, 2013, 341, 1240104 CrossRef PubMed.
- J. W. Shin, K. R. Spinler, J. Swift, J. A. Chasis, N. Mohandas and D. E. Discher, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18892–18897 CrossRef CAS PubMed.
- D. Bhattacharya, A. Czechowicz, A. G. Ooi, D. J. Rossi, D. Bryder and I. L. Weissman, J. Exp. Med., 2009, 206, 2837–2850 CrossRef CAS PubMed.
- T. Lapidot and I. Petit, Exp. Hematol., 2002, 30, 973–981 Search PubMed.
- K. Chatterjee, S. Lin-Gibson, W. E. Wallace, S. H. Parekh, Y. J. Lee, M. T. Cicerone, M. F. Young and C. G. Simon Jr., Biomaterials, 2010, 31, 5051–5062 CrossRef CAS PubMed.
- J. Klein-Nulend, A. D. Bakker, R. G. Bacabac, A. Vatsa and S. Weinbaum, Bone, 2013, 54, 182–190 CrossRef CAS PubMed.
- A. Mammoto, K. M. Connor, T. Mammoto, C. W. Yung, D. Huh, C. M. Aderman, G. Mostoslavsky, L. E. Smith and D. E. Ingber, Nature, 2009, 457, 1103–1108 CrossRef CAS PubMed.
- K. M. Stroka and H. Aranda-Espinoza, Blood, 2011, 118, 1632–1640 CrossRef CAS PubMed.
- R. S. Taichman, M. J. Reilly and S. G. Emerson, Blood, 1996, 87, 518–524 CAS.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed.
- A. J. Hodge and J. A. Petruska, in Aspects of Protein Structure, ed. G. N. Ramachandran, Academic Press, London, 1963, pp. 189–300 Search PubMed.
- R. Glass, M. Möller and J. P. Spatz, Nanotechnology, 2003, 14, 1153–1160 Search PubMed.
- J. P. Spatz, S. Mössmer, C. Hartmann and M. Möller, Langmuir, 2000, 16, 407–415 CrossRef CAS.
- T. Lohmuller, D. Aydin, M. Schwieder, C. Morhard, I. Louban, C. Pacholski and J. P. Spatz, Biointerphases, 2011, 6, MR1–M12 CrossRef PubMed.
- J. Blummel, N. Perschmann, D. Aydin, J. Drinjakovic, T. Surrey, M. Lopez-Garcia, H. Kessler and J. P. Spatz, Biomaterials, 2007, 28, 4739–4747 CrossRef PubMed.
- S. V. Graeter, J. Huang, N. Perschmann, M. Lopez-Garcia, H. Kessler, J. Ding and J. P. Spatz, Nano Lett., 2007, 7, 1413–1418 CrossRef CAS PubMed.
- M. Leiss, K. Beckmann, A. Giros, M. Costell and R. Fassler, Curr. Opin. Cell Biol., 2008, 20, 502–507 CrossRef CAS PubMed.
- E. A. Cavalcanti-Adam, D. Aydin, V. C. Hirschfeld-Warneken and J. P. Spatz, HFSP J., 2008, 2, 276–285 Search PubMed.
- E. A. Cavalcanti-Adam, A. Micoulet, J. Blummel, J. Auernheimer, H. Kessler and J. P. Spatz, Eur. J. Cell Biol., 2006, 85, 219–224 CrossRef CAS PubMed.
- E. A. Cavalcanti-Adam, T. Volberg, A. Micoulet, H. Kessler, B. Geiger and J. P. Spatz, Biophys. J., 2007, 92, 2964–2974 CrossRef CAS PubMed.
- V. C. Hirschfeld-Warneken, M. Arnold, A. Cavalcanti-Adam, M. Lopez-Garcia, H. Kessler and J. P. Spatz, Eur. J. Cell Biol., 2008, 87, 743–750 CrossRef CAS PubMed.
- M. Arnold, E. A. Cavalcanti-Adam, R. Glass, J. Blummel, W. Eck, M. Kantlehner, H. Kessler and J. P. Spatz, ChemPhysChem, 2004, 5, 383–388 CrossRef CAS PubMed.
- M. Arnold, V. C. Hirschfeld-Warneken, T. Lohmuller, P. Heil, J. Blummel, E. A. Cavalcanti-Adam, M. Lopez-Garcia, P. Walther, H. Kessler, B. Geiger and J. P. Spatz, Nano Lett., 2008, 8, 2063–2069 Search PubMed.
- M. J. Dalby, N. Gadegaard and R. O. Oreffo, Nat. Mater., 2014, 13, 558–569 CrossRef CAS PubMed.
- Y. Sun, C. S. Chen and J. Fu, Annu. Rev. Biophys., 2012, 41, 519–542 Search PubMed.
- J. E. Frith, R. J. Mills and J. J. Cooper-White, J. Cell Sci., 2012, 125, 317–327 Search PubMed.
- S. Oh, K. S. Brammer, Y. S. Li, D. Teng, A. J. Engler, S. Chien and S. Jin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2130–2135 Search PubMed.
- E. K. Yim, S. W. Pang and K. W. Leong, Exp. Cell Res., 2007, 313, 1820–1829 CrossRef CAS PubMed.
- J. Jiang and E. T. Papoutsakis, Adv. Healthcare Mater., 2013, 2, 25–42 Search PubMed.
- K. Ma, C. K. Chan, S. Liao, W. Y. Hwang, Q. Feng and S. Ramakrishna, Biomaterials, 2008, 29, 2096–2103 CrossRef CAS PubMed.
- J. Suo, D. E. Ferrara, D. Sorescu, R. E. Guldberg, W. R. Taylor and D. P. Giddens, Atertio. Thromb. Vasc. Biol., 2007, 27, 346–351 Search PubMed.
- C. Christophis, I. Taubert, G. R. Meseck, M. Schubert, M. Grunze, A. D. Ho and A. Rosenhahn, Biophys. J., 2011, 101, 585–593 CrossRef CAS PubMed.
- M. Hanke, I. Hoffmann, C. Christophis, M. Schubert, V. T. Hoang, A. Zepeda-Moreno, N. Baran, V. Eckstein, P. Wuchter, A. Rosenhahn and A. D. Ho, Biomaterials, 2014, 35, 1411–1419 CrossRef CAS PubMed.
- Y. Liu, T. Q. Liu, X. B. Fan, X. H. Ma and Z. F. Cui, J. Biotechnol., 2006, 124, 592–601 Search PubMed.
- L. K. Nielsen, Annu. Rev. Biomed. Eng., 1999, 1, 129–152 CrossRef CAS PubMed.
- A. Janowska-Wieczorek, M. Majka, J. Ratajczak and M. Z. Ratajczak, Stem Cells, 2001, 19, 99–107 Search PubMed.
- R. R. Kay, P. Langridge, D. Traynor and O. Hoeller, Nat. Rev. Mol. Cell Biol., 2008, 9, 455–463 CrossRef CAS.
- R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS.
- N. Di Maggio, E. Piccinini, M. Jaworski, A. Trumpp, D. J. Wendt and I. Martin, Biomaterials, 2011, 32, 321–329 CrossRef CAS PubMed.
- J. Lee and N. A. Kotov, Small, 2009, 5, 1008–1013 CrossRef CAS PubMed.
- T. Mortera-Blanco, A. Mantalaris, A. Bismarck, N. Aqel and N. Panoskaltsis, Biomaterials, 2011, 32, 9263–9270 CrossRef CAS PubMed.
- I. Kurth, K. Franke, T. Pompe, M. Bornhauser and C. Werner, Integr. Biol., 2009, 1, 427–434 RSC.
- I. Kurth, K. Franke, T. Pompe, M. Bornhauser and C. Werner, Macromol. Biosci., 2011, 11, 739–747 Search PubMed.
- M. P. Lutolf, R. Doyonnas, K. Havenstrite, K. Koleckar and H. M. Blau, Integr. Biol., 2009, 1, 59–69 RSC.
- M. P. Lutolf, P. M. Gilbert and H. M. Blau, Nature, 2009, 462, 433–441 CrossRef CAS PubMed.
Footnotes |
| † Human CD34+ cells. |
| ‡ Murine mononuclear cells from bone marrow. |
| § Murine Lin− Sca1+ cKit+ cells (LSK cells). |
| ¶ Murine Lin− Sca1+ cells. |
| || Murine Lin− cKit+ cells. |
| ** Human Lin− mononuclear cells from umbilical cord blood. |
| †† Human Lin−, CD34+, CD38+ cells. |
| ‡‡ Human mononuclear cells from umbilical cord blood. |
| §§ Murine Lin− Sca1+ cKit+ CD150+ cells. |
| ¶¶ Human CD133+ cells. |
| This journal is © The Royal Society of Chemistry 2014 |