Peptide decoration of nanovehicles to achieve active targeting and pathology-responsive cellular uptake for bone metastasis chemotherapy†
Xuli
Wang‡
*a,
Ye
Yang‡
b,
Huizhen
Jia
c,
Wanjian
Jia
a,
Scott
Miller
a,
Beth
Bowman
a,
Jun
Feng
*c and
Fenghuang
Zhan
*b
aDivision of Radiobiology, School of Medicine, University of Utah, 729 Arapeen Drive, Rm 2334, Salt Lake City, 84108, USA. E-mail: xuli.wang@utah.edu
bDepartment of Internal Medicine, Division of Hematology, Oncology, and Blood & Marrow Transplant, University of Iowa, Iowa City, IA 52242, USA. E-mail: fenghuang-zhan@uiowa.edu
cKey Laboratory of Biomedical Polymers of Ministry of Education & Department of Chemistry, Wuhan University, Wuhan 430072, China. E-mail: fengjun@whu.edu.cn
First published on 14th April 2014
Abstract
To improve bone metastasis chemotherapy, a peptide-conjugated diblock copolymer consisting of a chimeric peptide, poly(ethylene glycol) and poly(trimethylene carbonate) (Pep-b-PEG-b-PTMC) is fabricated as a drug carrier capable of bone-seeking as well as pathology-responsive charge reversal to ensure effective cellular uptake at the lesion sites. The chimeric peptide CKGHPGGPQAsp8 consists of an osteotropic anionic Asp8, a cathepsin K (CTSK)-cleavable substrate (HPGGPQ) and a cationic residue tethered to the polymer chain. Pep-b-PEG-b-PTMC can spontaneously self-assemble into negatively charged nanomicelles (∼75 nm). As to the model drug of doxorubicin, Pep-b-PEG-b-PTM shows 30.0 ± 1% and 90.1 ± 2% for the loading content and loading efficiency, respectively. High bone binding capability is demonstrated with 66% of Pep-b-PEG-b-PTMC micelles were able to bind to hydroxyl apatite, whereas less than 15% of Pep-free micelles were bound to hydroxyl apatite. The nanomicelles exhibit a negative-to-positive charge conversion from −18.5 ± 1.9 mV to 15.2 ± 1.8 mV upon exposure to CTSK, an enzyme overexpressed in bone metastatic microenvironments. Such a pathology-responsive transition would lead to remarkably enhanced cellular uptake of the nanomicelles upon reaching lesion sites, thus improving the drug efficacy as verified by the in vitro cytotoxicity assay and the in vivo study with the myeloma-bearing 5TGM1 mice model.
1. Introduction
Most patients with cancer die not because of the tumor in the primary site, but rather because it spread to other sites of which the bone is the most common organ to be affected.1,2 Patients with bone metastases generally cannot be treated curatively, e.g., only 20% of patients with breast cancer remain alive for five years after the diagnosis of bone metastases; more than 550![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people die annually with bone metastases.1,3 Effective chemotherapy of bone metastases has therefore remained an urgent challenge for cancer treatments.4
000 people die annually with bone metastases.1,3 Effective chemotherapy of bone metastases has therefore remained an urgent challenge for cancer treatments.4
The surface decoration of drug nanovehicles (NVs) with bone-targeting moieties is among the most used strategies to improve the chemotherapy against bone metastases.5 Given the remarkable feature of skeletal tissue rich in apatite (Ca10(PO4)6), many anionic agents with high apatite binding affinity including bisphosphonates (BPs) and oligopeptides (Asp6, Asp8 and Glu8) have been purposely incorporated into NVs for the sake of bone-seeking action.5–7 However, the treatment efficacies achieved by those bone-targeting NVs are still far from satisfactory; some of them were even less efficacious than the unmodified counterparts. The marked deviation from the expectation is thought to be associated with the dilemma that the attachment of anionic agents to NVs inevitably restricts cellular translocation due to the charge repulsion by the cell membranes.4–6 In turn it is difficult for the drugs loaded into NVs to enter cancerous cells to elicit therapeutic action. In contrast, despite the easy intracellular internalization, the applications of positively charged NVs suffer a lot from rapid in vivo clearance and non-specific uptake by healthy tissues resulting from the strong interaction with oppositely charged blood components and the cell membranes. Taking those into account, the solution to bone-targeted chemotherapy may eventually rely on the rational design of anionic bone-seeking NVs which are simultaneously capable of undergoing negative-to-positive charge transition that mostly occurs in bone metastatic sites.
One notable physicochemical feature of bone metastases is the predominant over-expression of several proteases including matrix metalloproteinase, urokinase plasminogen activator, and cathepsins (cathepsins K, L, and B).8 In particular, the development of cancer metastases in bones is found to be highly correlated with the expression level of the acid-activated cathepsin K enzyme (CTSK). The bone metastatic lesions express even dramatically higher level of CTSK compared to primary tumor and soft tissue metastases.9 The strong dependence of bone metastases on the CTSK level makes CTSK able to serve as a prognostic indicator for the diagnosis of bone metastases and as a therapeutic target for disease treatments.10–12 This pathology-dependent feature of CTSK overexpression evokes our interest to exploit CTSK as the biological stimulus to locally trigger the charge reversal of anionic bone-seeking NVs upon reaching bone metastatic sites.13–16
We have herein designed a bone-seeking, CTSK-responsive charge-reversal NV system for efficient chemotherapy of bone metastases. This delivery system was constructed on the core–shell nanomicelles self-assembled from a triblock copolymer (Pep-b-PEG-b-PTMC) composed of a peptide segment with the sequence CKGHPGGPQAsp8, poly(ethylene glycol) and poly(trimethylene carbonate). The chimeric peptide segment consisted of three main functional modules: the anionic Asp8 is responsible for the direction and accumulation of NVs into the skeletal tissues, the HPGGPQ serves as the CTSK-cleavable substrate,8 and the remaining cationic residue is chemically conjugated to the PEG shell of the micelles. In this way, the combination of the targeting function of Asp8 and the stealth effect of the PEG shell would help the drug-loaded Pep-b-PEG-b-PTMC nanomicelles prolong the circulation time and minimize the undesirable cellular uptake by healthy cells. Of special note is that the bone metastatic microenvironment featured with CTSK overexpression would locally stimulate the enzymatic cleavage of HPGGPQ moieties, leaving the cationic peptide residue on the shell periphery. This pathology-responsive charge reversal transition is expected to largely contribute to the selectively efficient internalization of nanomicelles into the neighboring cancerous cells (Scheme 1), thus improving the therapeutic efficacy. In principle, this strategy of bone-seeking, pathology-responsive charge-reversal NVs may be also applicable to other malignant diseases of skeletal tissue if the development of those diseases involves overexpression of certain enzymes. To verify our hypothesis, an anticancer drug DOX was used as the drug model and the in vitro and in vivo properties of the Pep-b-PEG-b-PTMC nanomicelles were comparatively explored under the CTSK-rich and CTSK-absent conditions.
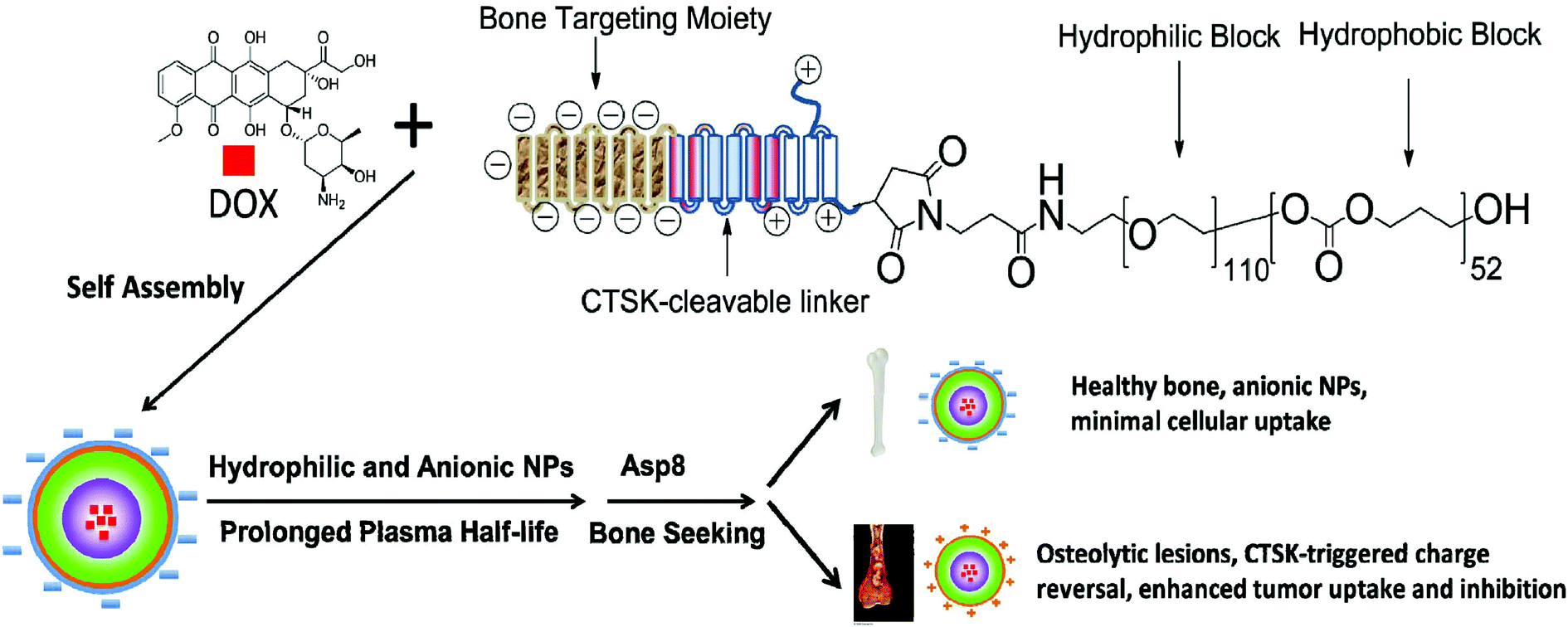 | ||
| Scheme 1 Schematic illustration of nanotherapeutic formation and its mechanism of action in vivo for bone metastasis treatment. | ||
2. Experimental section
2.1. Materials
Monomethyl terminated polyethylene glycol monoamine (MeO-PEG113-OH, Mw = 5000 Da) was purchased from Sigma-Aldrich. It was dried by azeodistillation of anhydrous benzene prior to use. Heterofunctional PEG with monomaleimide and monohydroxyl terminus (Mal-PEG110-OH, Mw = 5000 Da) was purchased from Laysan Bio (Arab, AL) and dried under high vacuum. Trimethylene carbonate (TMC) was prepared as previously described.17 It was recrystallized from a THF–ether mixture, and further purified via sublimation prior to use. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) was distilled from CaH2 under dry N2 prior to use. The N-(3,5-trifluoromethyl)phenyl-N-cyclohexylthiourea (TU) catalyst was prepared as described elsewhere.18 Stannous octoate (Sn(Oct)2, Sigma) was purified according to a method described in the literature.19 Anhydrous solvents were purchased from Sigma-Aldrich and stored over molecular sieves (4 Å). Doxorubicin was purchased from Selleck Inc. (Houston, TX). Cy5.5 was purchased from Lumiprobe (Russia). Cathepsin K was purchased from EMD Biosciences. Hydroxyapatite (HA) was purchased from Bio-Rad (Bio-Gel HTP, DNA grade; Hercules, CA). 1-Hydroxybenzotriazole (HOBt), O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU), Rink amide resin, NovaSyn TGT alcohol resin and all of the amino acids for solid phase peptide synthesis were purchased from Novabiochem (CA). All other chemical reagents were available commercially and used as received unless otherwise noted.2.2. Peptide synthesis
ISOLUTE column reservoirs (Charlottesville, VA) were used for the solid phase synthesis. The functional peptide with the sequence of CKGHPGGPQAsp8 was synthesized by a standard solid phase peptide synthesis (SPPS) procedure by using the Rink amide resin solid support and HBTU/HOBt as coupling reagents. Coupling efficiencies were monitored by the Kaiser ninhydrin test. The peptide was cleaved from the resin by treatment with the TFA–EDT–H2O–TIBS (94/2.5/2.5/1) mixture as a scavenger. The crude was precipitated with cold ethyl ether and purified by preparative RP-HLPC. The purity of the peptide was checked by analytical RP-HPLC and the molecular weight was confirmed by MALDI-TOF mass spectrometry, MS (m/z, [M]+): 1800.68 (calculated for C68H97N21O35S); 1800.54 (found).2.3. Synthesis of the MPEG-b-PTMC di-block copolymer (MPEG-b-PTMC)
HO-PEG113-OMe (5 g, 1 mmol), TMC (5.1 g, 50 mmol) and TU (380.6 mg, 2.5 mmol, 5 mol% of TMC monomer) were dissolved in 25 mL dichloromethane (DCM), and then DBU (380.6 mg, 2.5 mmol, 5 mol% of TMC monomer) was added. The reaction was performed under a N2 atmosphere for 24 hours. The reaction mixture was concentrated under a reduced pressure and the product was obtained by precipitation in ether. The residue was re-dissolved in a minimal amount of DCM and then poured into cold ether to precipitate the product; total yield: 8.6 g (85.2%).2.4. Synthesis of the maleimide-terminated PEG-b-PTMC polymer (Mal-PEG-b-PTMC)
Mal-PEG110-b-PTMC52 was synthesized by ring-opening polymerization using Mal-PEG110-OH (0.5 g) as the initiator, Sn(Oct)2 as the catalyst and anhydrous toluene as the solvent. The reaction was performed at 100 °C under a N2 atmosphere for 16 hours. The reaction mixture was concentrated under a reduced pressure and the product was obtained by precipitation in ether. The residue was re-dissolved in a minimal amount of THF and then poured into cold ether to precipitate the product. Yield: 0.72 g (71.3%).2.5. Synthesis of the peptide-b-PEG-b-PTMC copolymer (peptide-b-PEG-b-PTMC)
Peptide (CKGHPGGPQAsp8, 50 mg, 27.8 μmol) was dissolved in PBS buffer (pH = 7.2) and treated with tris(2-carboxyethyl)phosphine (TCEP, 40 μmol) for 2 hours under a N2 atmosphere. After that, 213 mg of Mal-PEG-b-PTMC in 5 mL of dimethylacetamide (DMAc) was added. The reaction was performed at room temperature overnight. The reaction mixture was then subject to dialysis against water (MWCO = 6–8 kDa, Spectrumlabs) at 4 °C for 24 hours. The solution was further purified using Microsep™ Centrifugal Devices with the Omega™ Membrane (MWCO = 10 kDa, Pall Life Sciences). The final product was recovered and dried by lyophilization. Yield: 151 mg (57.5%).2.6. NMR characterization
The MERCURY 400 MHz spectrometer was used for recording 1H NMR spectra to determine the structure and composition of the block copolymers. Deuterium oxide (D2O), deuterated chloroform (CDCl3) and dimethyl sulfoxide (DMSO-d6) were used as the solvents as noted for NMR measurements.2.7. Determination of critical micelle concentration (CMC)
Critical micelle concentrations of the copolymers were estimated by a fluorescence spectroscopy method using pyrene as the fluorescence probe.20 A predetermined amount of pyrene solution in acetone was added into a series of volumetric flasks, and the acetone was then evaporated completely. A series of copolymer solutions of different concentrations ranging from 1.0 × 10−5 to 1.0 mg mL−1 were added to the bottles, whereas the concentration of pyrene in each flask was fixed at a constant value (6.0 × 10−7 mol L−1). The excitation spectra were recorded at 20 °C on a Shimadzu RF-5301PC spectrofluorophotometer with the detection wavelength at 390 nm and a slit width of 3 nm. The intensity ratio of the bands at 340 and 338 nm (I340/I338) as a function of the logarithm of the concentration of copolymers was plotted and the CMC value was taken as the intersection of the tangents to the horizontal line of the intensity ratio with relatively constant values and the diagonal line with rapid increased intensity ratio.2.8. Preparation of self-assembled nanoparticles
mg of each polymer was dissolved in 1 mL of DMSO, followed by adding a predetermined amount of DOX·HCl and two molar equivalents of triethylamine (TEA) and stirred at room temperature for 1 h. Then, 5 mL of double distilled water was added dropwise. After being stirred for an additional 1 h, the solution was dialyzed (MWCO = 6–8 kDa, Spectrumlabs) against water for 24 h. Drug-loaded NVs were dried via lyophilization.
2.9. Determination of drug loading capacity
The drug loading content was determined spectrophotometrically by measuring the absorption of the lyophilized sample which was dissolved in DMSO at a wavelength of 482 nm. The absorbance of DOX in nanoparticles was measured to determine the drug content in the solution using an established calibration curve with a known concentration of free DOX. The percentages of DLC (drug loading content) and DLE (drug loading efficiency) were calculated according to the following equations: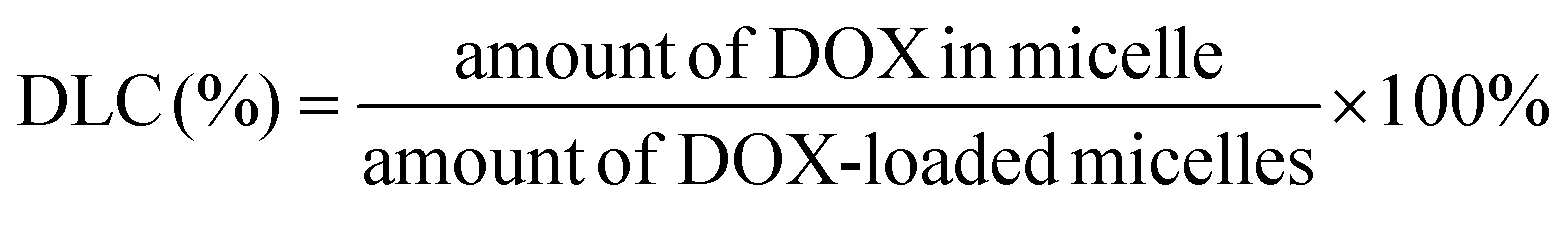
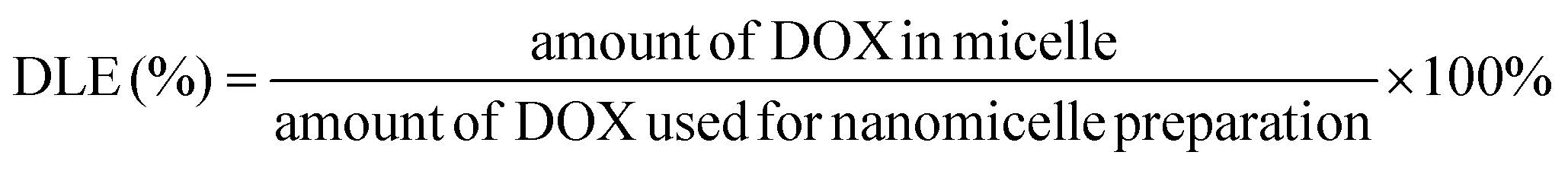
2.10. Drug release profile
Drug release experiments were performed in a buffer with a fixed ionic strength at 150 mM using saline. Appropriate pH was maintained with phosphate buffer (10 mM, pH 5.5 or 7.4) solutions. DOX-loaded micelle solution was diluted to 1 mg mL−1, and transferred into a dialysis membrane tubing (Spectra/Por, Float-A-Lyzer G2, MWCO = 8–10 kDa). The tubing was immersed in 100 mL of the release medium and kept in a horizontal laboratory shaker maintaining a constant temperature (37 °C) and stirring (100 rpm). Samples were periodically removed and replaced with the same volume of fresh medium. The amount of released DOX was analyzed with a spectrophotometer at 482 nm. The drug release studies were performed in triplicate for each of the samples.2.11. Dynamic light scattering (DLS)
The hydrodynamic size of the micellar nanoparticles was analyzed using a Brookhaven Instruments BI-200SM system equipped with a 5 mW helium neon laser with a wavelength output of 633 nm. The effective diameter and population distribution were computed from the diffusion coefficient. Measurements were made at 25 °C at an angle of 90°, and each sample was analyzed in triplicate. The micelle solution was filtered using a 0.45 μm membrane filter prior to measurements for all of the experiments.2.12. Binding affinity of nanoparticles to hydroxyapatite (HA)
The binding affinity of nanoparticles to hydroxyapatite was assessed with free Cy5.5, Cy5.5-loaded nanoparticles or Cy5.5-peptide conjugates.21 Cy5.5-peptide conjugates were obtained by direct conjugation of Mal-Cy5.5 (Lumiprobe, Russia) with the peptide via thiol–maleimide reaction. The samples were dissolved in phosphate buffered saline (pH 7.4) and then subjected to incubation with 1 mg of HA powder (HA, Bio-Gel HTP, DNA grade; BIO-RAD, Hercules, CA) in an eppendorf tube at RT. At predetermined time intervals, the incubation was stopped by centrifugation. The UV absorbance of the supernatant was monitored at a wavelength of 660 nm. Each sample was measured in duplicate.2.13. Cell culture
Myeloma cells (5TGM1 and ARP-1) and breast cancer cells (MCF-7) were cultured in RPMI1640 containing 10% heat-inactivated fetal calf serum (FCS), 4 mM L-glutamine (Gibco), penicillin (100 U mL−1), and streptomycin (100 μg mL−1) at 37 °C in a humidified atmosphere containing 5% CO2.2.14. Cell-proliferation assay
The cells were seeded in 96-well plates at 2000 cells per well in 100 μL of complete medium, and incubated at 37 °C in a 5% CO2 humidified atmosphere, followed by adding DOX-loaded nanoparticles or blank micelles (25 μL) at pre-determined concentrations. Following 48-hour incubation, 12.5 μL of PrestoBlue™ Cell Viability Reagent (Invitrogen) was added to cultured cells. After incubation for an additional 3 h, the absorbance of the solution was measured at 570 and 600 nm using a microplate reader (Molecular Device). The cell number was calculated according to the manufacturer's instructions, and the cell viability was normalized to that of cells without drug treatment.2.15. Confocal laser scanning microscopy (CLSM)
Confocal laser scanning microscopy was carried out using an Olympus FV-1000 laser scanning microscope operated with the FLUOVIEW software (Olympus, Tokyo, Japan). Images were produced using the lasers sequentially with a 20× objective lens.2.16. Biological evaluation of CTSK-pretreated nanoparticles
Cathepsin K (EMD Biosciences) was pre-incubated at 37 °C for 5 min to activate the enzyme in the active site, followed by addition of the nanoparticles. The nanoparticles were incubated with cathepsin K (150 nM) at 37 °C in acetate buffer (0.1 M, pH 5.5). The incubation mixture contained 0.15 μM of cathepsin K, 2.5 mM cysteine, and 5 mM EDTA. After CTSK pretreatment for 30 minutes, nanoparticles were recovered via ultrafiltration (MWCO = 100 kDa), and then subjected to zeta potential measurements (Malvern Zetasizer 3000), cell incubation for CLSM observation (Olympus FV-1000) or cytotoxicity evaluations. Nanoparticles were pretreated in the cleavage buffer, but in the absence of CTSK were used as the controls.2.17. In vivo test of nanotherapeutics
The preliminary test of DOX-loaded nanotherapeutics was carried out in a 5TGM1 myeloma cell-induced bone metastatic mouse model. Experiments were approved by the Institutional Animal Care and Use Committee of the University of Utah. To establish the mice with bone manifestations of metastatic cancers, 1 × 106 5TGM1 cells (in 100 μL PBS) were injected into each mouse through the tail veil and the tumors were allowed to grow for a week. The bone-targeted, CTSK-cleavable nanotherapeutics, as well as the corresponding non-targeted nanotherapeutics (neutral NVs), or free DOX were investigated. The doses of doxorubicin were kept equivalent in different groups. Another group with PBS was used as a negative control. Three C57BL/KaLwRij mice with 5TGM1 inoculations were used for each group. At the time of the experiments, the animal was injected with different therapeutic formulations or controls at a dose of 0.75 mg kg−1 twice a week. The tumor burden in the mice was monitored weekly by ELISA measurements of monoclonal immunoglobulin (Ig) in mouse sera. Survival was evaluated from the day of tumor inoculation until death.2.18. Statistical analysis
All measurements were conducted in duplicate or triplicate and expressed as mean ± standard deviation where indicated. Differences between the mean values were analyzed by two-sided Student's t test or one-way ANOVA. The Kaplan–Meier curve and log rank test were used for mouse survival. P values of <0.05 are considered statistically significant.3. Results and discussion
A peptide-conjugated diblock copolymer consisting of a peptide, poly(ethylene glycol) and poly(trimethylene carbonate) (Pep-b-PEG-b-PTMC) has been synthesized for the first time. PEG was used as the hydrophilic block because of its established role to help NVs escape from protein opsonization and macrophage uptake in the reticuloendothelial system. The rationale behind the choice of PTMC as the hydrophobic block was that PTMC can degrade with a relatively slower rate and have a weak inflammatory effect of degradation compounds, which is a marked advantage over polyesters and particularly attractive to sustained drug delivery application.22The synthesis of a peptide-conjugated diblock copolymer of Pep-b-PEG-b-PTMC was based on the efficient click reaction between the cysteine terminated Pep peptide and the maleimidevinyl terminated Mal-PEG-b-PTMC diblock copolymer, as illustrated in Fig. 1. Via the well-established organocatalytic (i.e. TU) ring-opening polymerization approach, a non-functional MPEG-b-PTMC copolymer could be readily prepared with a defined molecular weight and narrow molecular distribution when using MPEG-OH (Mw = 5000 Da) as the macroinitiator.23 This strategy was problematic, however, when it came to the synthesis of the maleimide-terminated copolymer of Mal-PEG-b-PTMC starting from Mal-PEG-OH. 1H NMR analysis revealed that the maleimidevinylic protons at σ = 6.74 ppm in the product disappeared. Apparently, the maleimide group might be very labile to super basic conditions, and failed to remain intact during the polymerization process. Alternatively, the Mal-PEG110-b-PTMC52, in which 52 refers to the repeating units of TMC, was successfully synthesized under mild conditions under Sn(Oct)2 catalysis (Fig. 1A). 1H NMR analysis demonstrated that the maleimidevinylic protons at σ = 6.74 ppm in the product remained available after polymerization (Fig. 2). The copolymer composition and number-averaged molecular weight of Mal-PEG110-b-PTMC52 were determined from the integral ratio of PTMC protons at σ = 4.21 ppm versus that of the PEG proton at σ = 3.64 ppm. For comparison purposes, the peptide-free methoxy-terminated MPEG113-b-PTMC50 was also prepared.
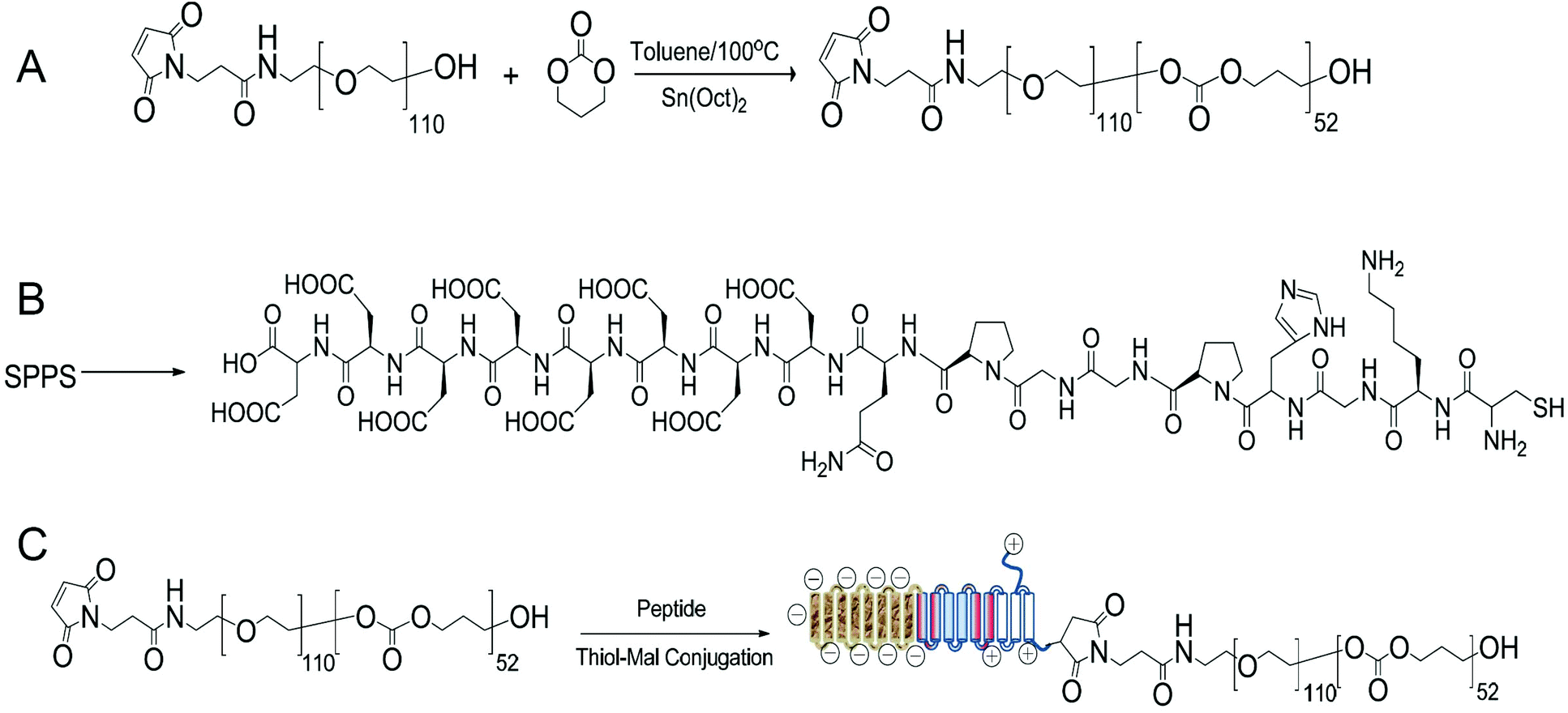 | ||
| Fig. 1 Synthetic procedure and chemical structure of Mal-PEG110-b-PTMC52 (A), the Pep peptide (B) and Pep-b-PEG110-b-PTMC52 (C). | ||
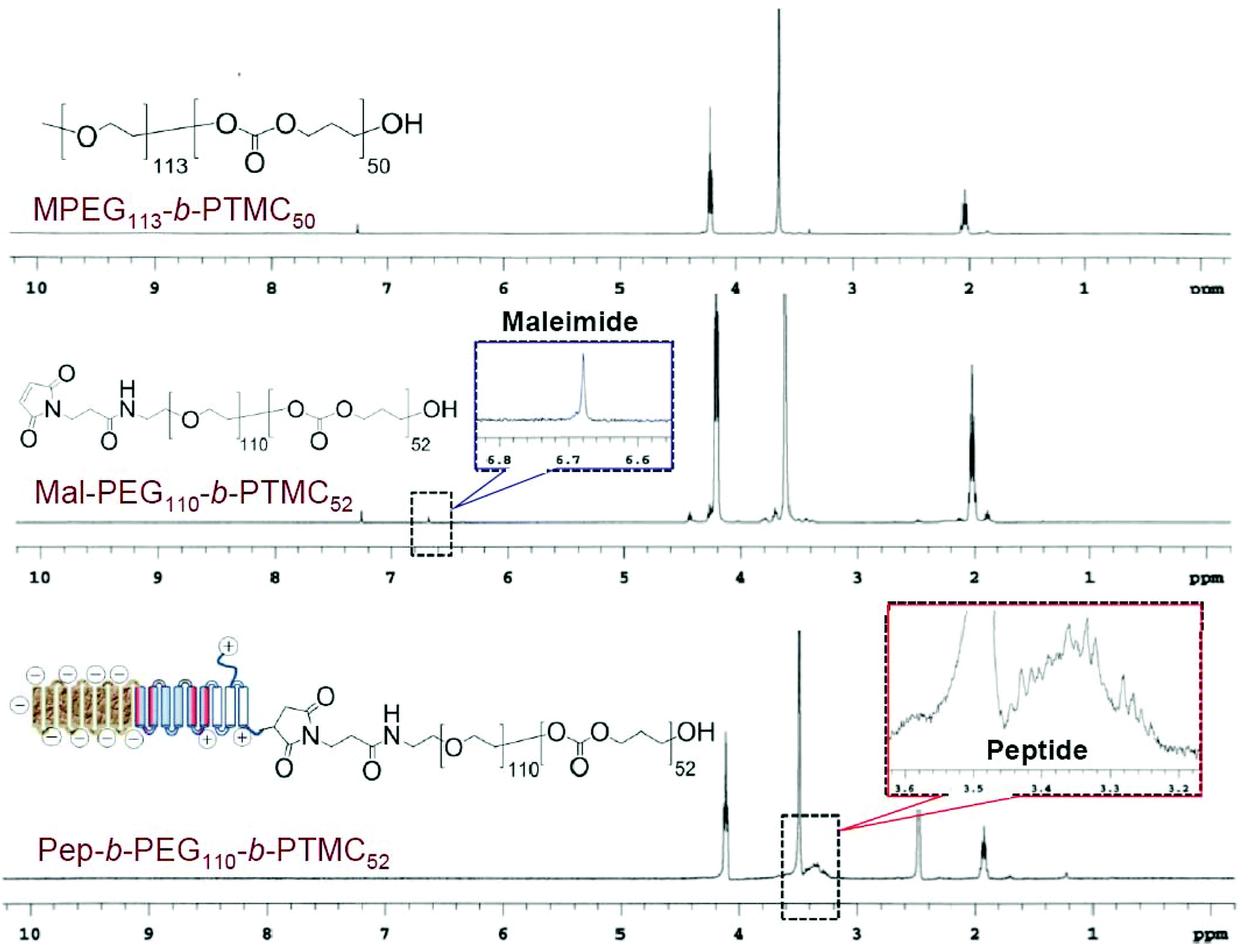 | ||
| Fig. 2 1H NMR spectra of MPEG113-b-PTMC50 (CDCl3), Mal-PEG110-b-PTMC52 (CDCl3) and Pep-b-PEG110-b-PTMC52 (DMSO-d6). | ||
Next, we synthesized the functional peptide with a specially designed sequence of CKGHPGGPQAsp8 based on the solid phase peptide synthesis (SPPS) technique (Fig. 1B). The purity (98%) and molecular weight (calculated: 1800.64, found: 1800.54) of the target peptide were confirmed by high-performance liquid chromatography (HPLC) and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry, respectively (Fig. S1†). Via click chemistry, the chemical conjugation of CKGHPGGPQAsp8 to Mal-PEG110-PTMC52 afforded the target copolymer PEP-b-PEG110-b-PTMC52 (Fig. 1C), which was purified by the dialysis method (MWCO = 10 kDa) followed by ultrafiltration treatment. Successful conjugation was verified by 1H NMR, which clearly showed the characteristic resonances of the peptide protons at σ = 3.1–3.6 ppm and complete disappearance of the maleimidevinylic signal at σ = 6.74 ppm (Fig. 2).
As expected, Pep-b-PEG-b-PTMC could readily self-assemble into the micelles in aqueous solution like many other amphiphilic copolymers,24 which was confirmed by the fluorescence assay using pyrene as the probe. According to the excitation spectra of pyrene at different polymer concentrations, the critical micelle concentration (CMC) of Pep-b-PEG110-b-PTMC52 was determined to be approximately 8.02 × 10−3 mg mL−1 (Fig. 3C). The mean hydrodynamic diameter of the polymeric micelles was measured by dynamic light scattering (DLS) to be 75 ± 10 nm, showing unimodal size distribution with PDI ∼ 0.1.
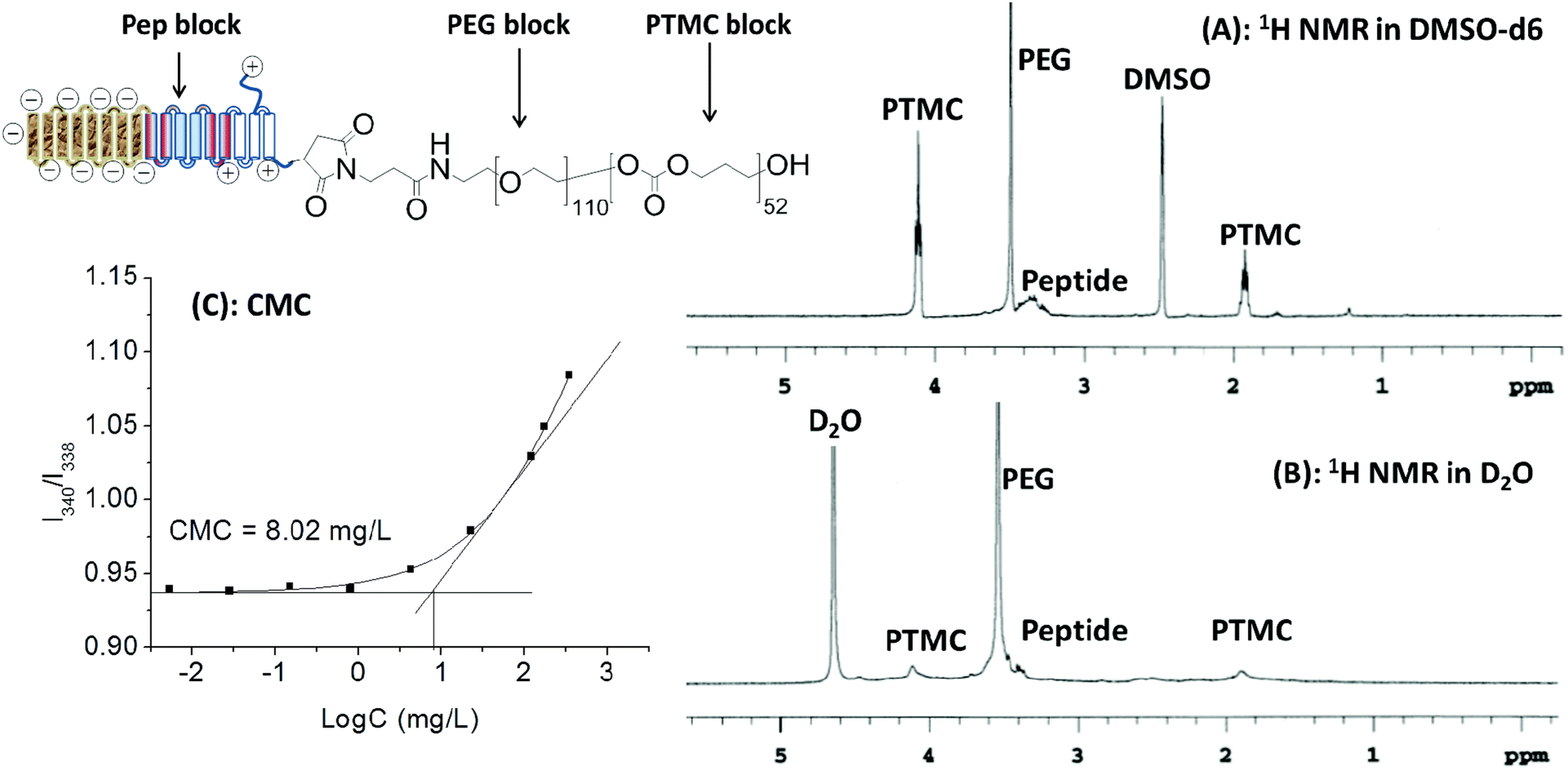 | ||
| Fig. 3 1H NMR spectra of Pep-b-PEG110-b-PTMC52 in DMSO-d6 (A) and D2O (B); CMC measurement by plotting of the I340/I338 ratio against the logarithm of the polymer concentration (C). | ||
In principle, the hydrophobic PTMC segments should localize in the inner core of nanomicelles while the peptide and PEG blocks form a corona-type shell due to their highly hydrophilic nature. This core–shell structure was verified by the marked difference between its 1H NMR spectra in DMSO (Fig. 3A) and D2O (Fig. 3B). All the characteristic signals could be detectable in DMSO-d6. Unlike this, the proton signals of the PTMC block were significantly suppressed and nearly disappeared in aqueous D2O but those from hydrophilic blocks remained prominent. Taken together, those characterizations demonstrated that Pep-b-PEG110-b-PTMC52 was able to readily form the NVs with the core–shell architecture in the aqueous medium.
The strong binding ability of Pep-b-PEG110-b-PTMC52 micelles to hydroxyapatite (HA), commonly used as a model mineral to mimic the bone tissue mineral, was examined to preliminarily evaluate the contribution of the peptide functionality to the bone-seeking efficacy.5–7 A near infra-red fluorescence (NIRF) Cy5.5 probe has been one of the frequently used fluorescence indicators due to its deeper tissue penetration capacity.25 Cy5.5 was encapsulated into the micellar cores by the dialysis method thus to quantitatively determine the binding affinities of the nanomicelles with and without the peptide functionality.
After incubation for 30 min, approximately 55% of the Pep-b-PEG-b-PTMC micelles from the solution were rapidly bound to HA. Further incubation for 24 hours led to a moderate increase of HA-binding up to about 66% (Fig. 4). In marked contrast, less than 15% of MPEG-b-PTMC micelles or free Cy5.5 were bound to HA across the entire incubation process, indicating minimal nonspecific binding to HA by the MPEG-b-PTMC micelles without the peptide functionality. The low-molecular-weight peptide-Cy5.5 conjugate exhibited a much stronger affinity to HA with complete binding after incubation for 30 min. Those results correlated well with the reported findings that modification with osteotropic Asp8 would dramatically facilitate the specific binding towards HA-rich bone tissue.26–28 Though the outer PEG shell with steric hindrance may somewhat restrain the complete binding of Pep-b-PEG-b-PTMC micelles to HA, they still displayed dramatically enhanced HA affinities over Pep-free MPEG-b-PTMC micelles after all.
 | ||
| Fig. 4 Binding capacity of various Cy5.5 formulations to the bone mineral HA. | ||
Typically, the decoration of bone-targeting anionic Asp8 to NVs would disfavor the cellular uptake process due to the charge repulsion by the cell membrane, thus impairing the chemotherapy efficacy.29 Within the structure of Pep-b-PEG-b-PTMC, a CTSK cleavable substrate (HPGGPQ)25,30 was specially incorporated to link Asp8 and the rest of the block copolymer. By virtue of such a CTSK-cleavable spacer, it was speculated that the bone metastatic microenvironment featured with CTSK overexpression would induce the enzymatic chain cleavage and the following removal of Asp8 moieties from the shell periphery of the nanomicelles. Of particular interest, this CTSK-induced transition may reverse the surface charge and thus enhance the cellular uptake of the nanomicelles, which was attributed to the remaining cationic peptide residues after Asp8 removal. To verify this speculation, the zeta potentials of polymeric micelles were measured in the presence and absence of CTSK, respectively. Following 30 min treatment with CTSK, the zeta potential of Pep-b-PEG-b-PTMC micelles had an instantly marked promotion from −18.5 ± 1.9 mV to 15.2 ± 1.8 mV (Fig. 5). Nevertheless, the control experiment without CTSK treatment afforded nearly no variation of the zeta potential. Apparently, it is just CTSK exposure that allowed the charge reversal of the Pep-b-PEG-b-PTMC micelles.
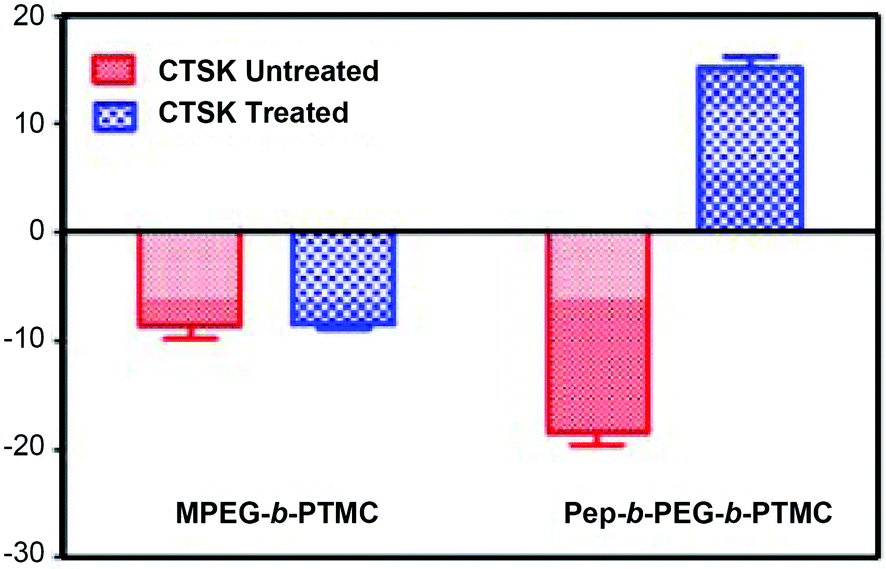 | ||
| Fig. 5 Change in the zeta potential of Pep-b-PEG-b-PTMC micelles upon co-incubation with 150 nM CTSK for 30 minutes. | ||
For comparison, the CTSK-responsiveness of the zeta potential was also tested for MPEG-b-PTMC micelles with the identical CTSK treatment. No apparent variation was found upon CTSK exposure and the micellar zeta potential remained almost constant within a narrow range from −8.6 ± 2.1 mV to −8.4 ± 0.7 mV. Those results pointed out that the introduction of the CKGHPGGPQAsp8 functionality indeed played a dominant role in the CTSK-induced negative-to-positive charge conversion of Pep-b-PEG-b-PTMC micelles.
To evidence the contribution of CTSK-responsive charge reversal to the intracellular internalization, confocal laser scanning microscopy (CLMS) was used to investigate the cellular uptake of polymeric micelles under the conditions with or without CTSK treatment. The nanomicelles assembled from Pep-b-PEG-b-PTMC were loaded with the Cy5.5 fluorescence indicator, followed by incubation with MCF-7 cells in the presence or absence of CTSK. Following 30 min incubation, the MCF-7 cells were subjected to CLMS observation. As shown in Fig. 6, very weak red fluorescence can be detected in the cells co-incubated with Pep-b-PEG-b-PTMC micelles in the medium free of CTSK, indicating that few fluorescence-labeled micelles were taken up by MCF-7 cells under this condition. The poor cellular uptake of untreated micelles was reasonably expected, because it was difficult for the negatively charged NVs to be intracellularly internalized owing to the charge repulsion effect. However, upon CTSK exposure, significant fluorescence enhancement was observed from the CLSM images. This was ascribed to the cationic nature of the micelles resulting from CTSK-induced charge-reversal, which was known to favor the approach of NVs to the negatively charged cell membranes and thus facilitate their cellular uptake. For the control of MPEG-b-PTMC micelles, it is not strange that a minimal difference was observed between the CLSM images obtained before and after CTSK treatment (data not shown).
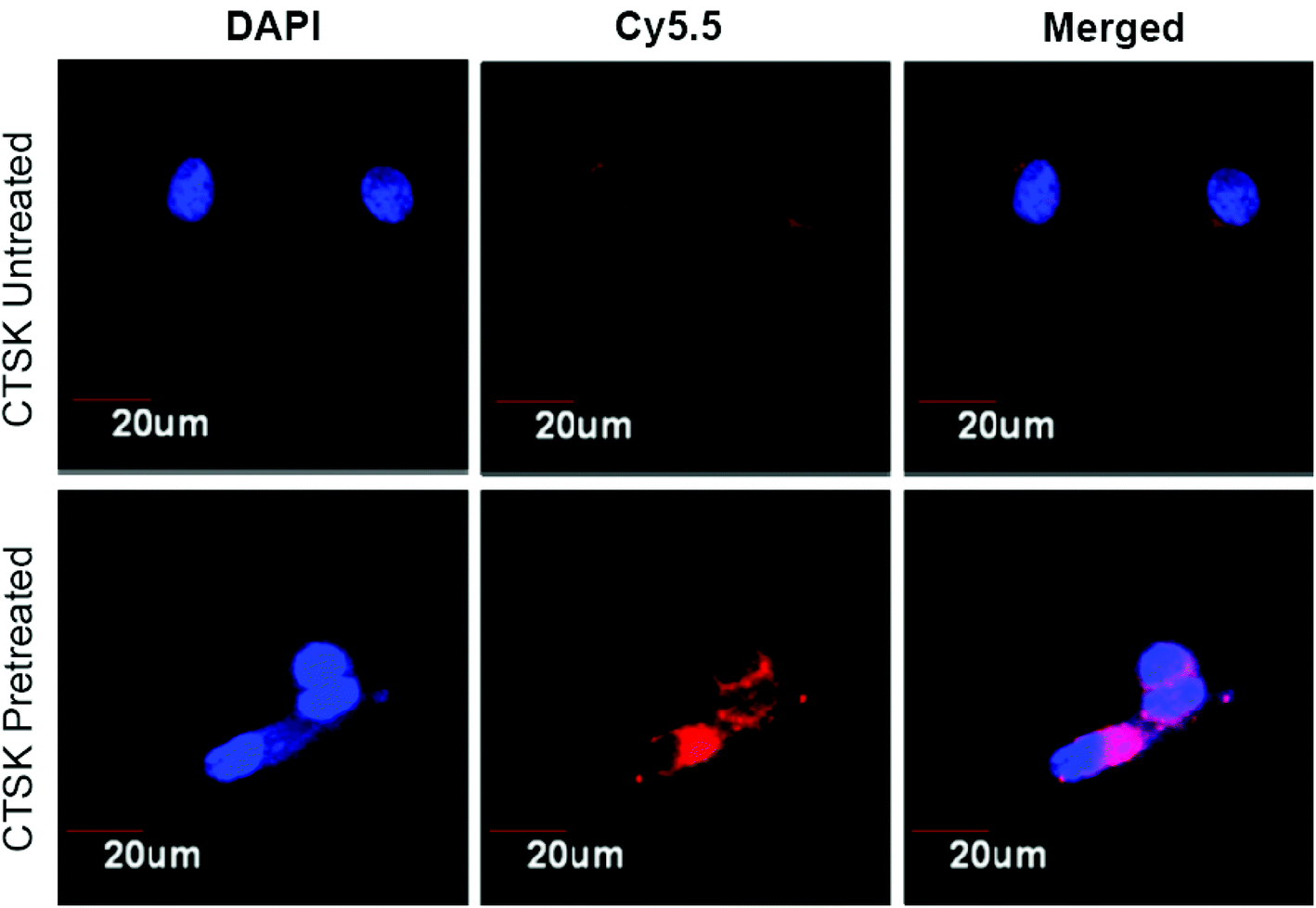 | ||
| Fig. 6 CLSM images of MCF-7 cells incubated with Pep-b-PEG-b-PTMC micelles with or without CTSK treatment. | ||
The CTSK-dependent cellular uptake of Pep-b-PEG-b-PTMC nanomicelles seemed very attractive in terms of the chemotherapy potential for bone metastases. It can be noted that those nanomicelles would be hardly taken up by the cells in the normal tissues due to the lack of CTSK expression; once the nanoparticles were guided and accumulated in CTSK-rich environments (i.e., CTSK over-expressed bone metastases lesions), however, the transition to cationic NVs would considerably facilitate the entry into the neighboring tumor cell and thus allow the effective treatment with lower systematical biotoxicity.
To further explore the potential of Pep-b-PEG-b-PTMC as a drug carrier, an anticancer drug DOX was used as a model drug to investigate the drug loading and release properties. Poorly water-soluble DOX was encapsulated into the micelles of Pep-b-PEG-b-PTMC or MPEG-b-PTMC by the dialysis method. Typical DOX-loading capacity for mPEG-b-PTMC micelles was determined as 7.5 ± 0.5% and 22.7 ± 1.5% for DLC (drug loading content) and DLE (drug loading efficiency), respectively. Dramatic increases of DLC (30.0 ± 1%) and DLE (90.1 ± 2%) were observed when Pep-b-PEG-b-PTMC was used as the drug carrier. Such a high loading efficiency may be the result of the charged nature of Pep-b-PEG-b-PTMC, for which the outer PEG chains extend more freely and more aggregation numbers per micelle are required to ensure the hydrophilic–hydrophobic balance in the aqueous medium. Thus the DOX drug molecules enter the micelle interior more readily and the larger core is more capable of containing more drug cargos. This is consistent with the finding reported in the literature that charged NVs generally possess high DOX-loading capacity.13,31–33 The representative example is that the highest drug loading (47% w/w) of DOX-loaded micelles reported to date was achieved when anionic polymeric glutamic acid (PGA) was used as a building block within PTMC-b-PGA.31 The in vitro drug release study was performed under a simulated physiological condition (pH 7.4). Fig. 7A compares the drug release profiles between the free DOX and the DOX loaded in Pep-b-PEG-b-PTMC micelles. The results suggested that DOX-loaded micelles exhibited a moderately rapid release in the first stage (around 10%) followed by a sustained release period. The sustained DOX release could be attributed to the hydrophobic interaction of drug molecules with the hydrophobic core of the polymeric micelles.
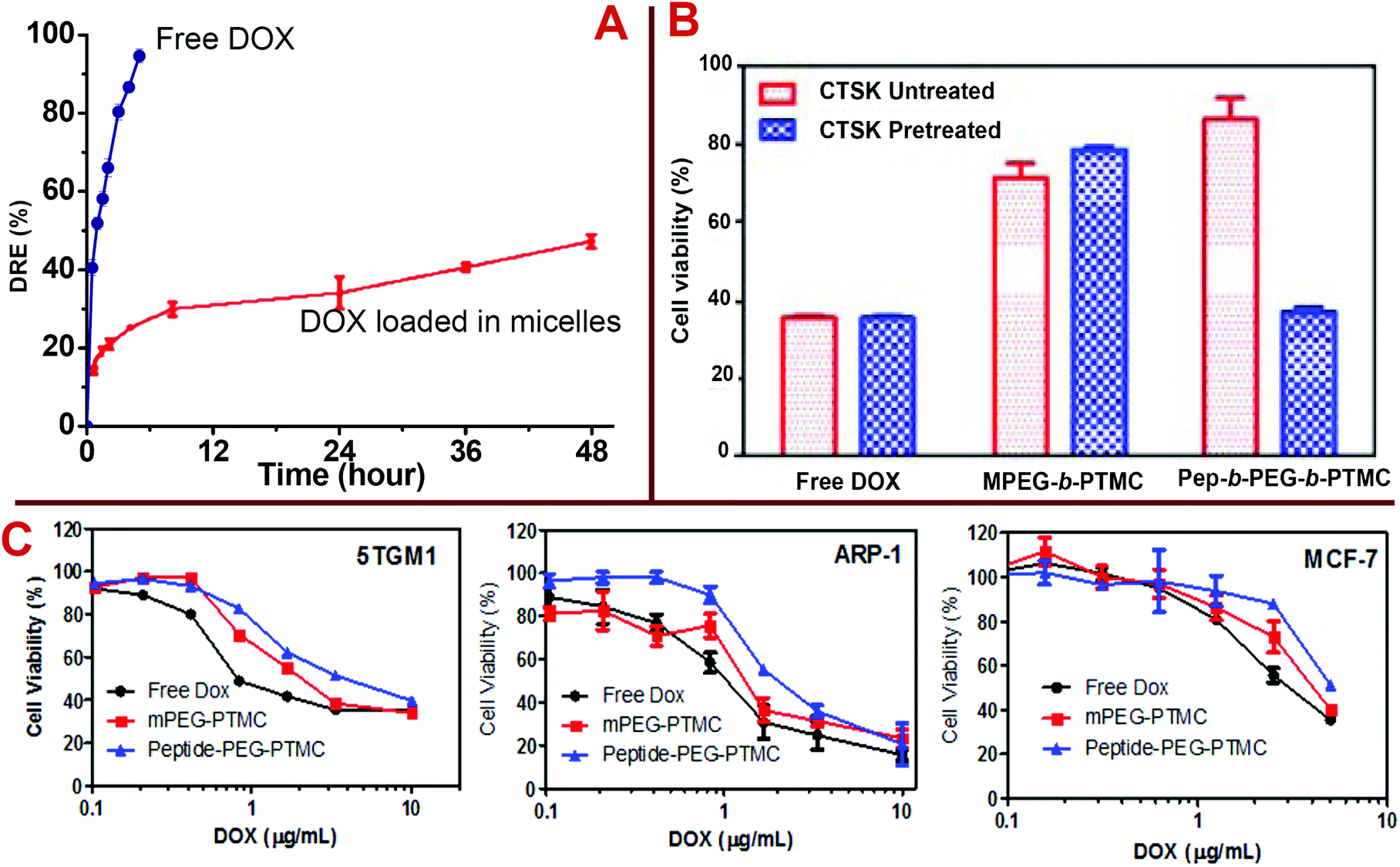 | ||
| Fig. 7 (A) Cumulative DOX release from the polymeric micelles at pH = 7.4. (B) Cell viability of 5TGM1 cells after incubation with free DOX, DOX-loaded MPEG-b-PTMC nanomicelles, and DOX-loaded Pep-b-PEG-b-PTMC nanomicelles at the same DOX concentration of 1 μg mL−1. Formulations were pre-treated with (blue) or without (red) 150 nM CTSK for 30 minutes. (C) In vitro inhibition of 5TGM1, ARP-1 and MCF-7 cells after 48 h incubation with free DOX or DOX-loaded NPs at various doses; data are presented as the average ± standard deviation (n = 3). | ||
The in vitro cytotoxicity profiles of free DOX or DOX-loaded polymeric micelles were evaluated in the cultured cells including myeloma (the most frequent cancer type for bone metastases) suspension cell lines (5TGM1 and ARP-1) and a breast cancer (the second most frequent cancer type for bone metastases) adherent cell line (MCF-7) by the PrestoBlue™ (Invitrogen) assay. The PrestoBlue assay performed very well to quantitatively measure cell proliferation and to establish the relative viability of both suspension and adherent cells. As shown in Fig. 7C, free DOX exhibited a relatively higher toxic effect than that of both DOX-loaded Pep-free and Pep-functionalized polymeric micelles after 4 h culture. The lowered potency of DOX after encapsulation into the micelles was probably due to the sustained release pattern of DOX from the micelles and delayed drug efficacy in the cultured cells. Overall, DOX-loaded Pep-PEG-b-PTMC nanomicelles showed lower cytotoxicity than the Pep-free MPEG-b-PTMC micelles. Since the CTSK activity has strong association with the interactions between tumor cells and the bone microenvironment, to give more exact information about the influence of CTSK-responsive charge-reversal on the biological properties of Pep-b-PEG-b-PTMC micelles, the 5TGM1 cytotoxicity of the DOX-loaded micelles was comparatively explored after a short co-incubation period of 30 min under the conditions with or without CTSK treatment. At an equivalent DOX dose (1 μg mL−1), drug-loaded Pep-b-PEG-b-PTMC micelles showed lower capability to induce the cell death than the Pep-free MPEG-b-PTMC control. CTSK treatment resulted in significant improvement of the drug efficacy in the case of Pep-b-PEG-b-PTMC micelles (Fig. 7B), which was even comparable to the acute toxicity of free DOX. Noticeably, the CTSK-responsive improvement of toxic effect was not observed for free DOX or DOX-loaded mPEG-b-PTMC micelles. This marked contrast manifested a close tie between the CTSK responsiveness and the Pep functionality in the micelles, which contributed much to the enhanced drug efficacy. In combination with the result of intracellular internalization obtained from confocal images, it is naturally inferred that the anionic nature associated with the targeting Asp8 moieties would disfavor the intracellular internalization of the Pep-b-PEG-b-PTMC micelles and therefore impair the drug efficacy, whereas CTSK-responsive charge reversal may more or less overcome this biological barrier.
Despite the advance of drug delivery nanosystems for bone metastases, to the best of our knowledge, nearly no in vivo efficacy evaluation in animal studies have yet been reported. This could be in part ascribed to the limited availability of suitable detection means for the disease progression of bone metastases. Herein, a preliminary in vivo study was conducted to test the efficacy of DOX-loaded Pep-b-PEG-b-PTMC nanomicelles in a 5TGM1-induced myeloma-bearing mice model. Multiple myeloma is the most frequent cancer-induced osteolytic disease,1,2 which intends to invade skeletal tissues in a similar mechanism to cancer cells that interact with the bone marrow microenvironment.34 Likewise, the interactions between myeloma cells and osteoclasts yield substantial cathepsin K up-regulation, resulting in osteolytic lesions.35 Noticeably, the real-time tumor burden in 5TGM1 mice can be easily monitored by measuring the 5TGM1 idiotype (IgG2b) in mouse sera.36 The higher IgG2b level in sera, the bigger tumor in the body. This is a distinct advantage since the progression of other types of cancers can only be identified with advanced imaging modalities (μCT, X-ray, etc.).37 Myeloma-bearing 5TGM1 mice were established through inoculation of 5TGM1 cells (1 × 106 cells in 100 μL PBS) via tail veil injection, and the tumors were allowed to develop and grow for one week before initiating study treatments. The mice were injected twice weekly with different therapeutic formulations at an equivalent DOX dose (0.75 mg kg−1) or blank PBS, and the IgG2b level in sera was monitored by ELISA measurements.
As shown in Fig. 8A, the myeloma-bearing 5TGM1 mice group treated with DOX-loaded Pep-b-PEG-b-PTMC micelles showed prolonged survival rate in comparison with the control groups with the administration of DOX-loaded MPEG-b-PTMC micelles, free DOX or blank PBS. Since the tumor burden was positively correlated with the IgG2b level, the data in Fig. 8B distinctly indicated that Pep-b-PEG-b-PTMC/DOX nanotherapeutic was meanwhile more effective to slow down tumor growth than the other formulations with the equivalent DOX dose. Together with the in vitro results, the observed improvement of both in vivo systematic biosafety and drug efficacy manifested the superiority of our pathology-specific design by combining both the advantages of efficient DOX delivery to bone metastatic tissues and CTSK-responsive cellular uptake of DOX within lesion sites.
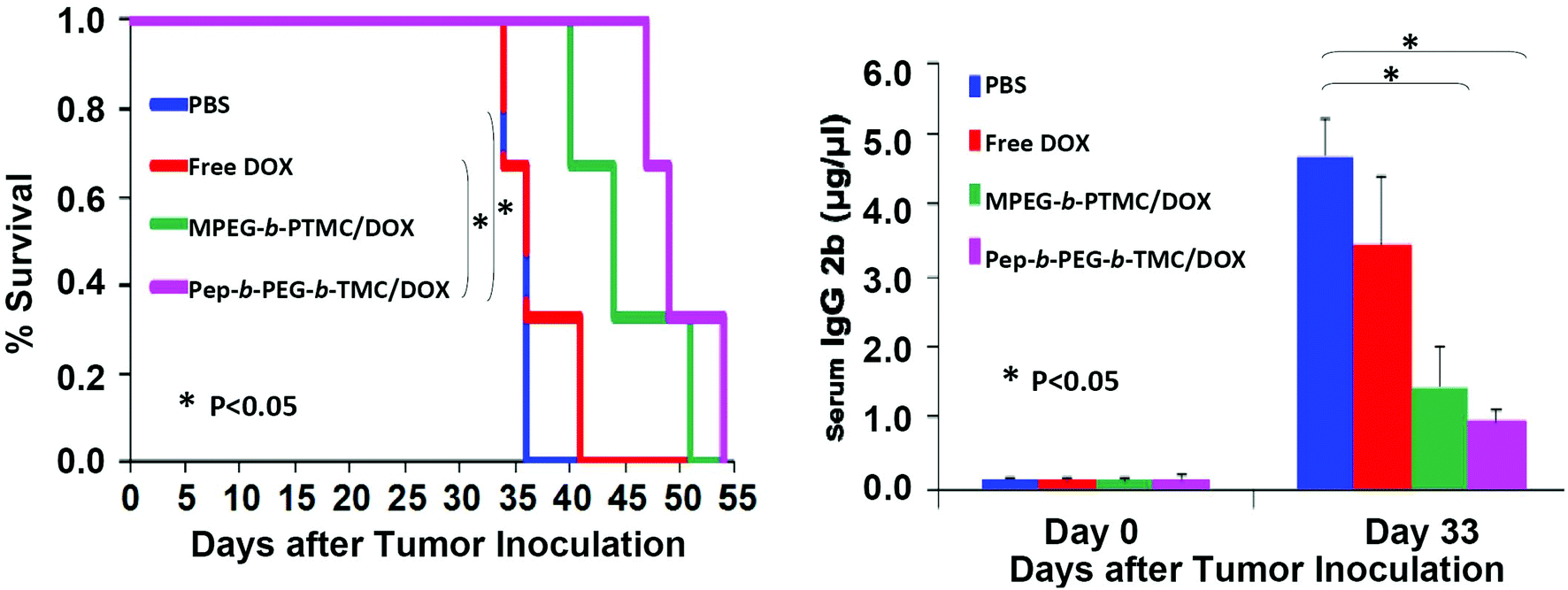 | ||
| Fig. 8 (A) Efficacy of free DOX or DOX-loaded nanomicelles in bone metastatic 5TGM1 mice bearing myeloma; (B) Kaplan–Meier survival curves of mice following tumor injection and treatments. Relative tumour burden in mice was determined by the IgG 2b level in sera. *P < 0.05 (n = 3). | ||
4. Conclusions
Cancer induced bone metastases often lead to substantial morbidity and mortality, which remain a significant therapeutic challenge for cancer treatment. In attempts to address this challenge, a peptide decoration strategy was proposed to render the NVs both the bone-seeking function and the charge-reversal capability in response to the pathological microenvironment. The nanomicelles self-assembled from the Pep-b-PEG-b-PTMC polymer showed strong apatite binding affinity and readily underwent the negative-to-positive charge reversal upon exposure to CTSK, an overexpressed enzyme in bone metastatic microenvironments. This unique characteristic led to dramatically enhanced cellular uptake within bone metastatic sites in a pathology-responsive manner, thus improving the drug efficacy of the loaded DOX in vitro and in vivo. Although our attempt presented herein for the enhanced bone metastasis chemotherapy is still in an initial stage, the obtained in vivo and in intro results are fairly encouraging. More attractively, the success of this strategy may open a new avenue to chemotherapy for the malignant diseases in bone tissue, including but not limited to bone metastases, provided that the development of therapy for those diseases involves overexpression of certain enzymes.Acknowledgements
This research was supported by NIH (R01CA166941, X. Wang), the Department of Defense (W81XWH1110307 and W81XWH1310240, X. Wang), the University of Utah CCTS Pilot Award (U59004183, X. Wang) from the NIH National Center for Research Resources (UL1RR025746), the TRP Award (6246-11, F. Zhan) from the Leukemia & Lymphoma Society, and the National Natural Science Foundation of China (grant no. 21374085, J. Feng).Notes and references
- G. D. Roodman, N Engl. J. Med., 2004, 350(16), 1655–1664 CrossRef CAS PubMed.
- L. S. Loftus, S. Edwards-Bennett and G. H. Sokol, Cancer Control, 2012, 19(2), 145–153 CAS.
- C. M. Bagi, Adv. Drug Delivery Rev., 2005, 57(7), 995–1010 CrossRef CAS PubMed.
- R. E. Coleman, Nat. Rev. Clin. Oncol., 2012, 9(2), 76–78 CrossRef CAS PubMed.
- D. Wang, S. C. Miller, P. Kopeckova and J. Kopecek, Adv. Drug Delivery Rev., 2005, 57(7), 1049–1076 CrossRef CAS PubMed.
- K. Miller, R. Erez, E. Segal, D. Shabat and R. Satchi-Fainaro, Angew. Chem., Int. Ed., 2009, 48(16), 2949–2954 CrossRef CAS PubMed.
- D. Wang, S. C. Miller, L. S. Shlyakhtenko, A. M. Portillo, X. M. Liu, K. Papangkorn, P. Kopekova, Y. Lyubchenko, W. I. Higuchi and J. Kopecek, Bioconjugate Chem., 2007, 18(5), 1375–1378 CrossRef CAS PubMed.
- Q. Zhao, Y. Jia and Y. Xiao, Biochem. Biophys. Res. Commun., 2009, 380(4), 721–723 CrossRef CAS PubMed.
- C. Le Gall, A. Bellahcene, E. Bonnelye, J. A. Gasser, V. Castronovo, J. Green, J. Zimmermann and P. Clezardin, Cancer Res., 2007, 67(20), 9894–9902 CrossRef CAS PubMed.
- R. N. Pearse, Clin. Breast Cancer, 2007, 8, S35–S45 CrossRef CAS.
- C. Le Gall, E. Bonnelye and P. Clezardin, Curr. Opin. Support. Palliat. Care, 2008, 2(3), 218–222 CrossRef PubMed.
- J. Sturge, M. P. Caley and J. Waxman, Nat. Rev. Clin. Oncol., 2011, 8(6), 357–368 CAS.
- J. Z. Du, T. M. Sun, W. J. Song, J. Wu and J. Wang, Angew. Chem., Int. Ed., 2010, 49(21), 3621–3626 CrossRef CAS PubMed.
- Y. Lee, T. Ishii, H. Cabral, H. J. Kim, J. H. Seo, N. Nishiyama, H. Oshima, K. Osada and K. Kataoka, Angew. Chem., Int. Ed., 2009, 48(29), 5309–5312 CrossRef CAS PubMed.
- Y. Lee, K. Miyata, M. Oba, T. Ishii, S. Fukushima, M. Han, H. Koyama, N. Nishiyama and K. Kataoka, Angew. Chem., Int. Ed., 2008, 47(28), 5163–5166 CrossRef CAS PubMed.
- P. S. Xu, E. A. Van Kirk, Y. H. Zhan, W. J. Murdoch, M. Radosz and Y. Q. Shen, Angew. Chem., Int. Ed., 2007, 46(26), 4999–5002 CrossRef CAS PubMed.
- T. Ariga, T. Takata and T. Endo, Macromolecules, 1997, 30(4), 737–744 CrossRef CAS.
- A. P. Dove, R. C. Pratt, B. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127(40), 13798–13799 CrossRef CAS PubMed.
- H. R. Kricheldorf, I. Kreiser-Saunders and A. Stricker, Macromolecules, 2000, 33(3), 702–709 CrossRef CAS.
- J. Z. Du, D. P. Chen, Y. C. Wang, C. S. Xiao, Y. J. Lu, J. Wang and G. Z. Zhang, Biomacromolecules, 2006, 7(6), 1898–1903 CrossRef CAS PubMed.
- H. Z. Pan, P. Kopeckova, D. Wang, J. Y. Yang, S. Miller and J. Kopecek, J. Drug Target, 2006, 14(6), 425–435 CrossRef CAS PubMed.
- J. Feng, X. Z. Zhang and R. X. Zhuo, Prog. Polym. Sci., 2012, 37(2), 211–236 CrossRef CAS PubMed.
- F. Nederberg, B. G. G. Lohmeijer, F. Leibfarth, R. C. Pratt, J. Choi, A. P. Dove, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2007, 8(1), 153–160 CrossRef CAS PubMed.
- L. Liu, K. Xu, H. Wang, P. K. Tan, W. Fan, S. S. Venkatraman, L. Li and Y. Y. Yang, Nat. Nanotechnol., 2009, 4(7), 457–463 CrossRef CAS PubMed.
- K. M. Kozloff, L. Quinti, S. Patntirapong, P. V. Hauschka, C. H. Tung, R. Weissleder and U. Mahmood, Bone, 2009, 44(2), 190–198 CrossRef CAS PubMed.
- M. B. Murphy, J. D. Hartgerink, A. Goepferich and A. G. Mikos, Biomacromolecules, 2007, 8(7), 2237–2243 CrossRef CAS PubMed.
- S. Kasugai, R. Fujisawa, Y. Waki, K. Miyamoto and K. Ohya, J. Bone Miner. Res., 2000, 15(5), 936–943 CrossRef CAS PubMed.
- D. Wang, S. Miller, M. Sima, P. Kopekova and J. Kopecek, Bioconjugate Chem., 2003, 14(5), 853–859 CrossRef CAS PubMed.
- K. Xiao, Y. Li, J. Luo, J. S. Lee, W. Xiao, A. M. Gonik, R. G. Agarwal and K. S. Lam, Biomaterials, 2011, 32(13), 3435–3446 CrossRef CAS PubMed.
- F. A. Jaffer, D. E. Kim, L. Quinti, C. H. Tung, E. Aikawa, A. N. Pande, R. H. Kohler, G. P. Shi, P. Libby and R. Weissleder, Circulation, 2007, 115(17), 2292–2298 CrossRef CAS PubMed.
- C. Sanson, C. Schatz, J. F. Le Meins, A. Soum, J. Thevenot, E. Garanger and S. Lecommandoux, J. Controlled Release, 2010, 147(3), 428–435 CrossRef CAS PubMed.
- C. Yang, J. Tan, W. Cheng, A. Attia, C. Tan, Y. Ting, A. Nelson, J. L. Hedrick and Y. Y. Yang, Nano Today, 2009, 5, 515–523 CrossRef PubMed.
- N. Wiradharma, Y. Zhang, S. Venkataraman, J. L. Hedrick and Y. Y. Yang, Nano Today, 2009, 4(4), 302–317 CrossRef CAS PubMed.
- J. A. Fowler, C. M. Edwards and P. I. Croucher, Bone, 2011, 48(1), 121–128 CrossRef PubMed.
- M. Hecht, I. von Metzler, K. Sack, M. Kaiser and O. Sezer, Exp. Cell Res., 2008, 314(5), 1082–1093 CrossRef CAS PubMed.
- B. O. Oyajobi, G. Franchin, P. J. Williams, D. Pulkrabek, A. Gupta, S. Munoz, B. Grubbs, M. Zhao, D. Chen, B. Sherry and G. R. Mundy, Blood, 2003, 102(1), 311–319 CrossRef CAS PubMed.
- T. J. Rosol, S. H. Tannehill-Gregg, B. E. LeRoy, S. Mandl and C. H. Contag, Cancer, 2003, 97(3), 748–757 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4bm00020j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |